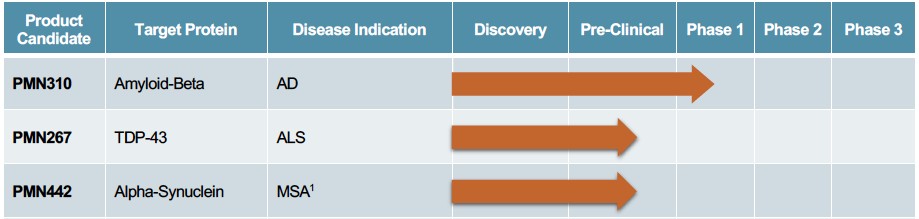
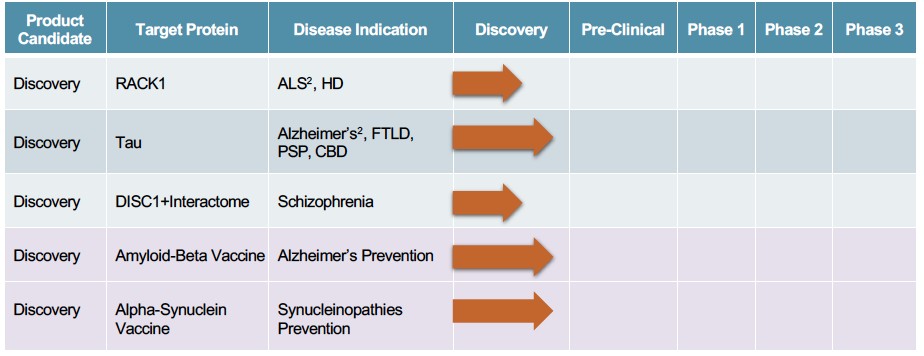
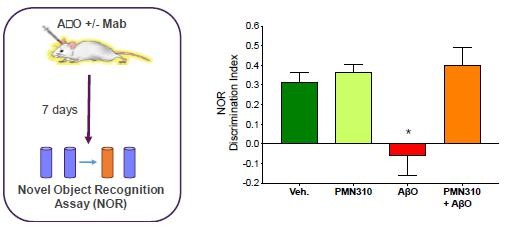
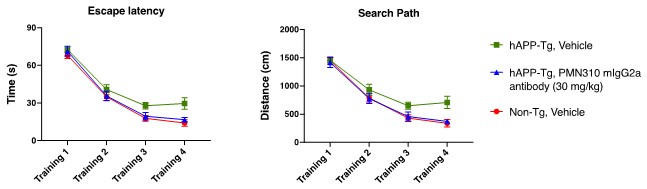
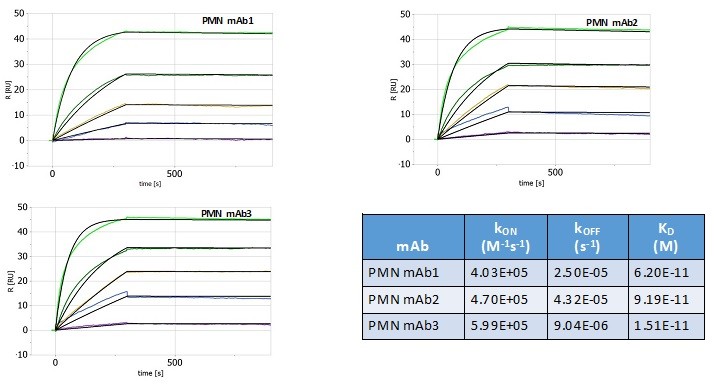
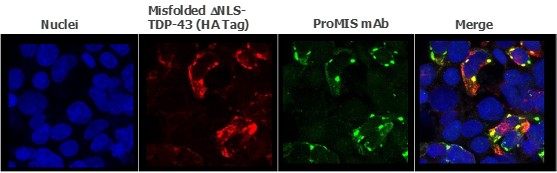
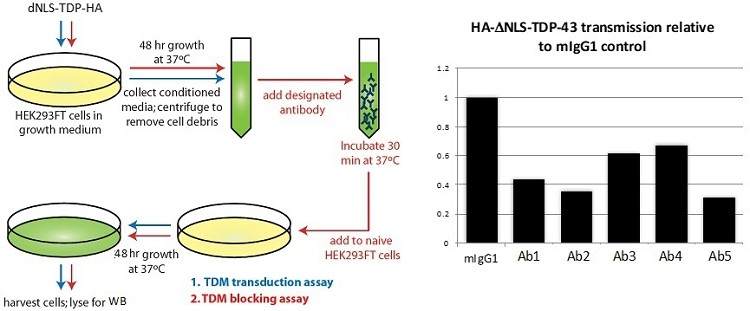
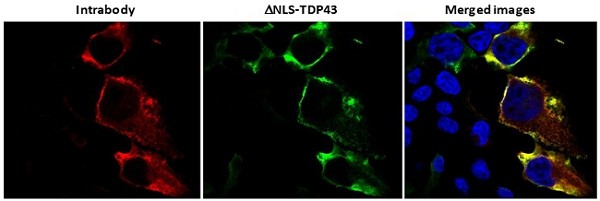
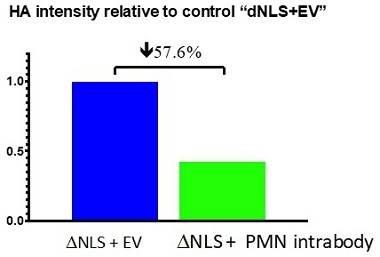
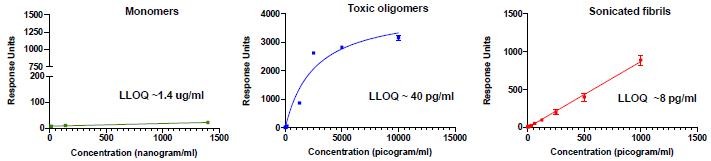
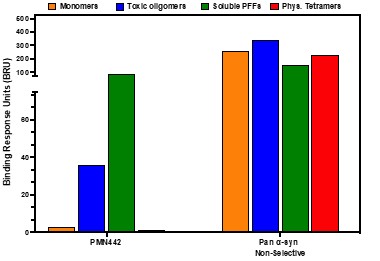
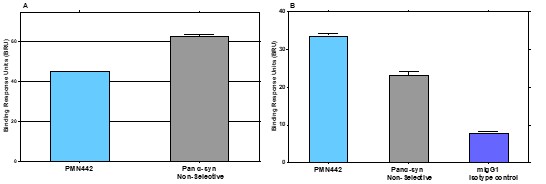
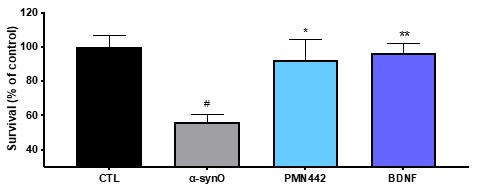
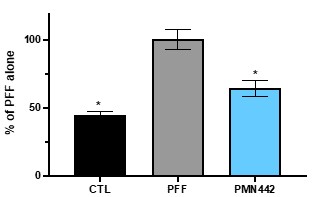
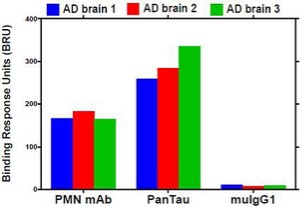
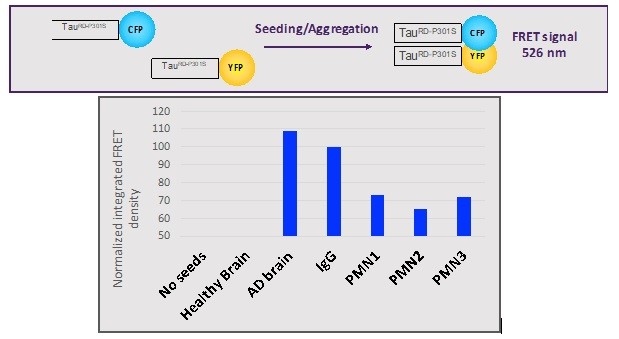
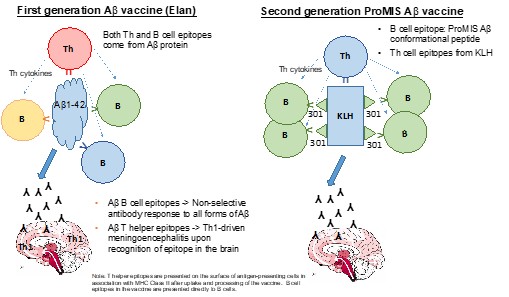
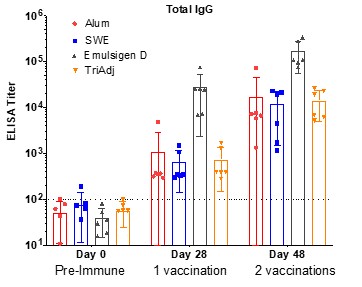
Human Capital Management
ProMIS seeks to hire qualified scientists and key employees as needed. As of December 31, 2023, the Company employed six full-time employees and one part-time employee. The remainder of the scientists and key personnel had consulting agreements with ProMIS.
Our future success depends on our ability to attract, develop and retain key personnel, maintain our culture, and ensure diversity and inclusion in our board, management and broader workforce. Our human resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and additional employees. The principal purposes of our equity incentive plans are to attract, retain and motivate selected employees, consultants and directors through the granting of stock-based compensation awards. As these areas directly impact our ability to compete and innovate, they are key focus areas for our board of directors and senior executives.
Corporate Structure
ProMIS Neurosciences Inc. was incorporated on January 23, 2004 under the name 4203801 Canada Inc. pursuant to the Canada Business Corporations Act (CBCA). The Company changed its name to Amorfix Life Sciences Ltd. on August 24, 2004 and to ProMIS Neurosciences Inc. effective July 8, 2015. On July 13, 2023, the Company continued its existence from a corporation incorporated under the CBCA into the Province of Ontario under the Business Corporations Act (Ontario) (OBCA) (Continuance). The Continuance was approved by the Company’s shareholders at the Company’s 2023 Annual Meeting of Shareholders held on June 29, 2023.
On June 21, 2022, the directors of the Company authorized a reverse share split of the issued and outstanding Common Shares in a ratio of 60:1, effective June 28, 2022 (the Reverse Share Split). All information included in this Annual Report on Form 10-K has been adjusted to reflect the Reverse Share Split. Unless otherwise stated herein, all share and per share numbers relating to the Company’s Common Shares prior to the effectiveness of the Reverse Share Split have been adjusted to give effect to the Reverse Share Split, including the consolidated financial statements and notes thereto. The Company’s Common Shares are listed on the Nasdaq Capital Market (Nasdaq) under the symbol, “PMN.”
Our head office is located at 1920 Yonge Street, Suite 200, Toronto, Ontario, Canada M4S 3E2 and our registered and records office is located at 1055 West Georgia Street, Vancouver, British Columbia, Canada V6E 4N7. Our telephone number is (416) 847-6898 and our website address is www.promisneurosciences.com. The information provided on our website is not part of this Annual Report on Form 10-K.
We own or have rights to various trademarks, service marks and trade names that we use in connection with the operation of our business. This Annual Report on Form 10-K may also contain trademarks, service marks and trade names of third parties, which are the property of their respective owners. Our use or display of third parties’ trademarks, service marks, trade names or products in this Annual Report on Form 10-K is not intended to, and does not imply a relationship with, or endorsement or sponsorship by us. Solely for convenience, the trademarks, service marks and trade names referred to in this Annual Report on Form 10-K may appear without the ®, ™ or SM symbols, but the omission of such references is not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable owner of these trademarks, service marks and trade names.
Unless the context indicates otherwise, references in this prospectus to the “Company,” “ProMIS,” “we,” “us,” “our,” and similar terms refer to ProMIS Neurosciences Inc. and its consolidated subsidiary.
Unless otherwise indicated, all references to “$” or “US$” in this Annual Report on Form 10-K refer to U.S. dollars, and all references to “C$” refer to Canadian dollars. Following the Company’s voluntary delisting from the Toronto Stock Exchange in July 2023, the Company reassessed its functional currency and determined that, as of July 1, 2023, its functional currency had changed from the C$ to the US$.
Available Information
We will make available on our website, free of charge, our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the