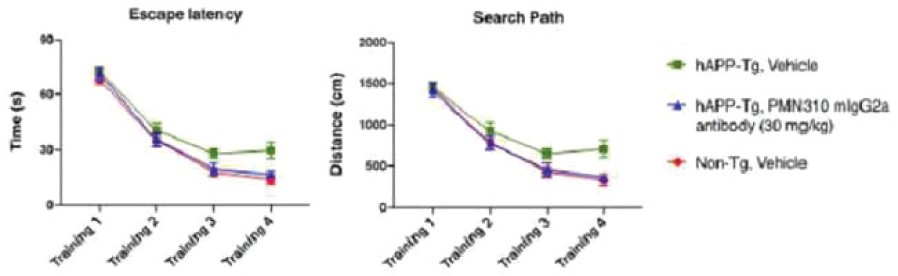
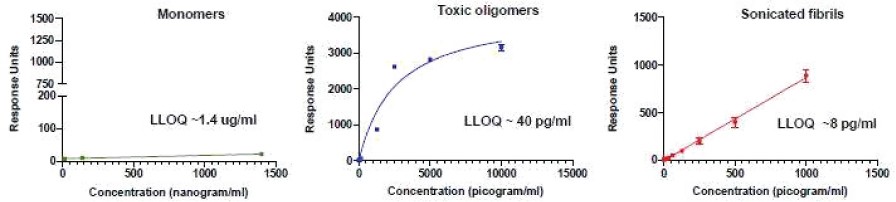
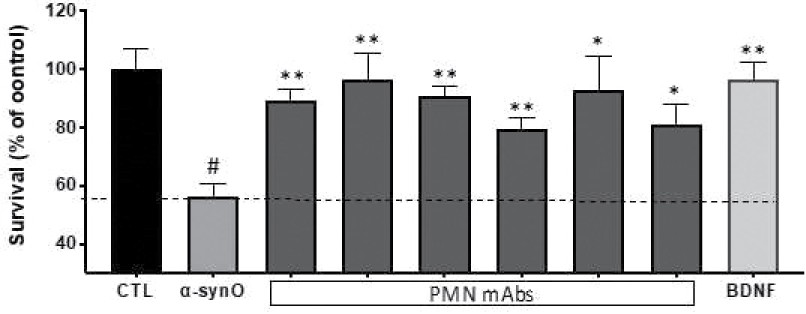
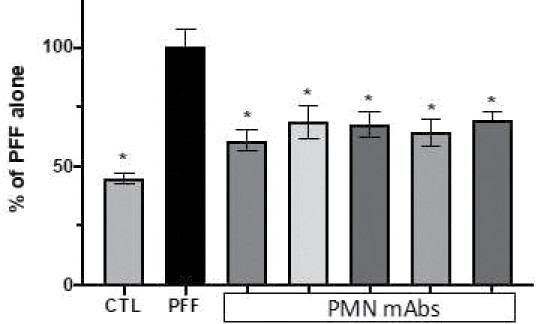
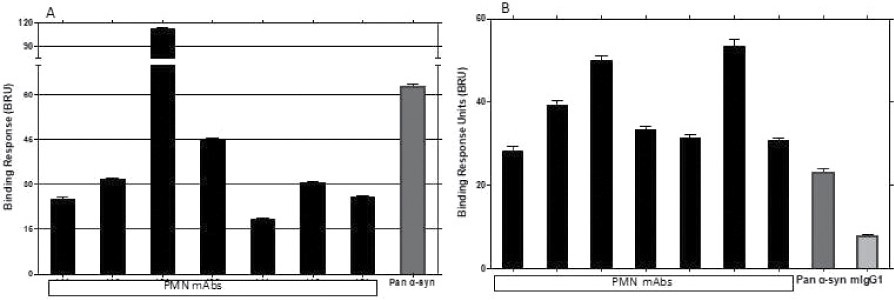
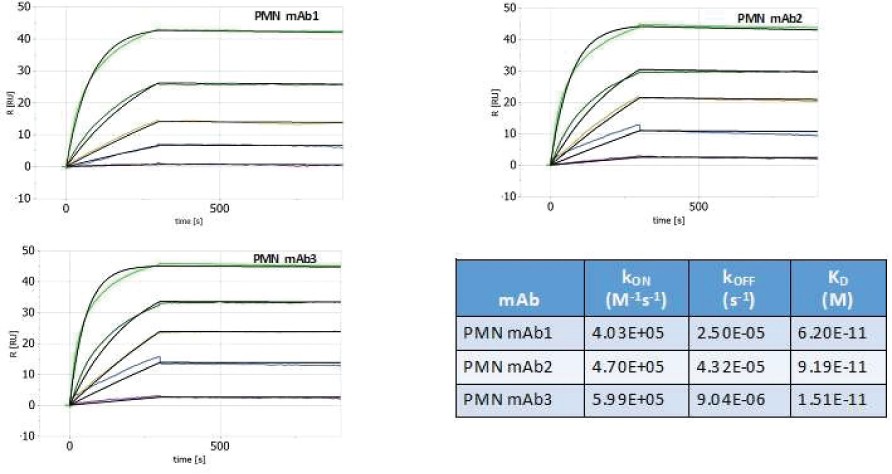
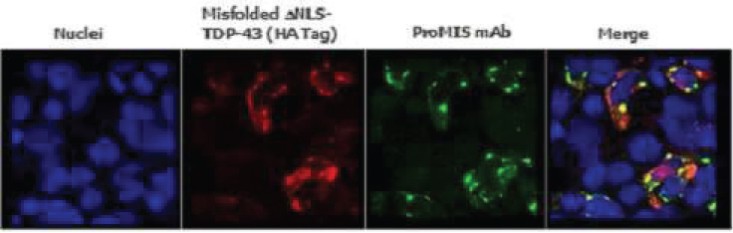
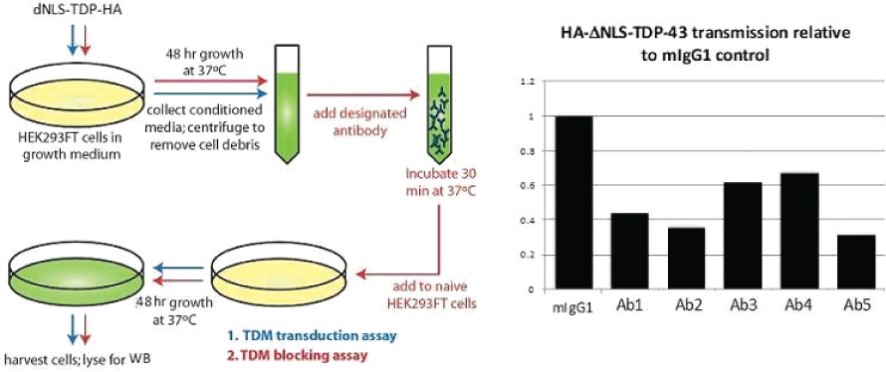
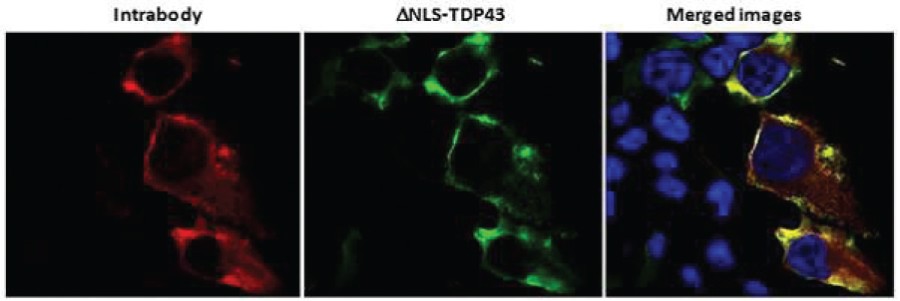
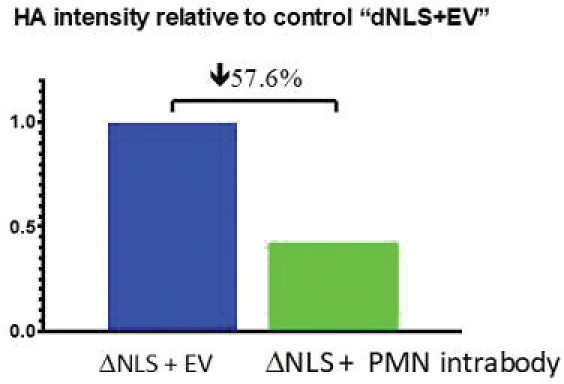
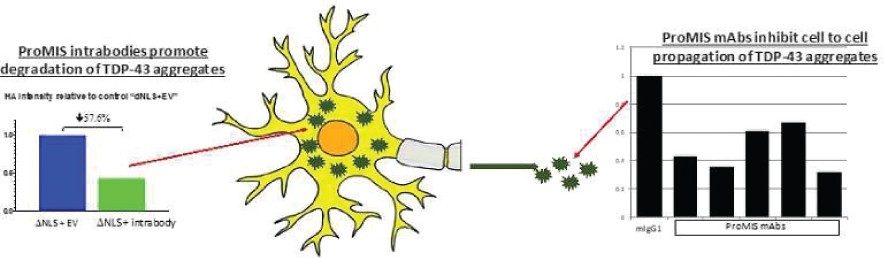
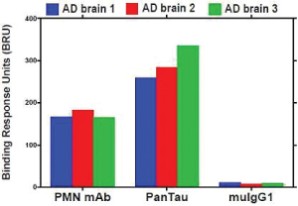
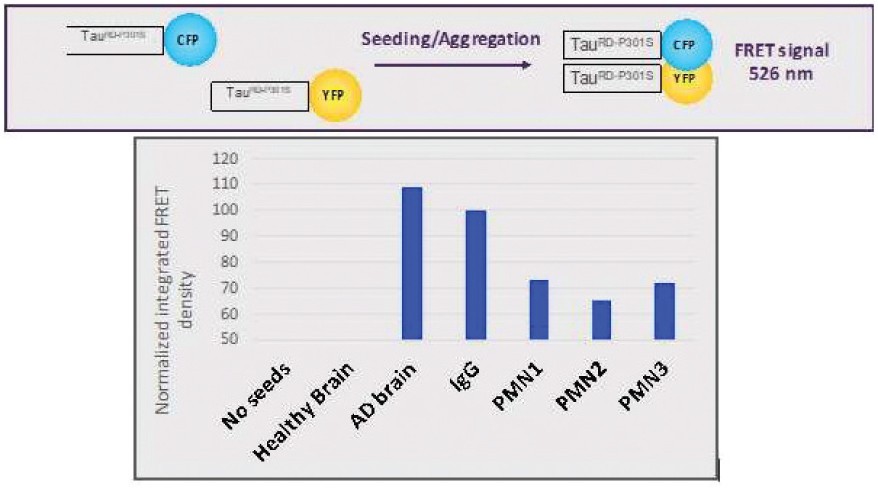
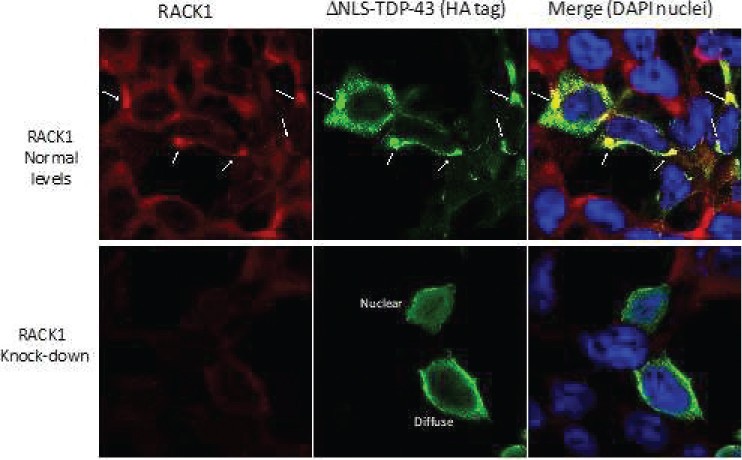
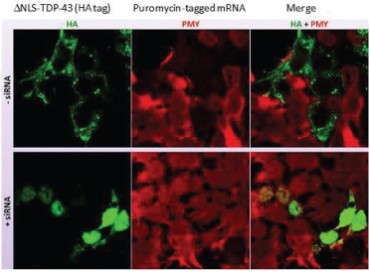
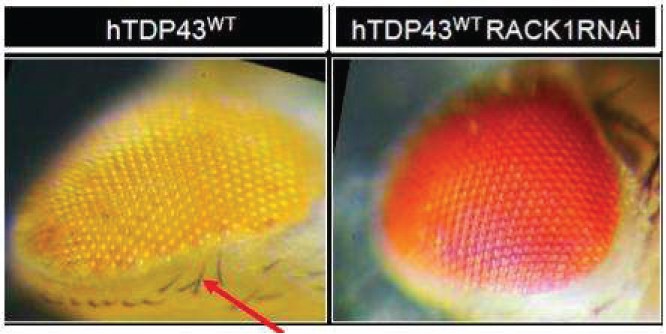
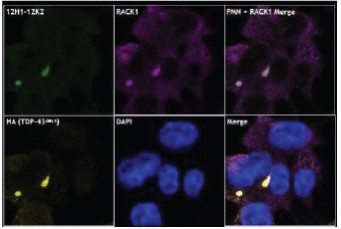
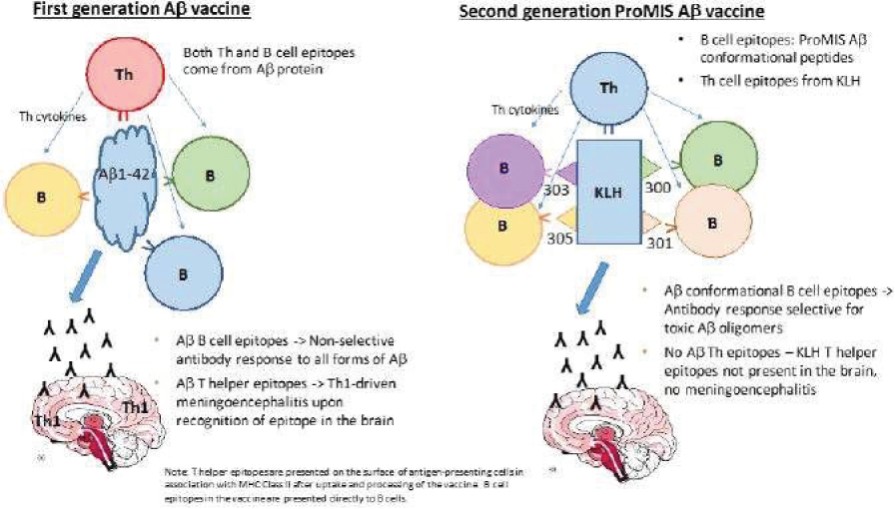
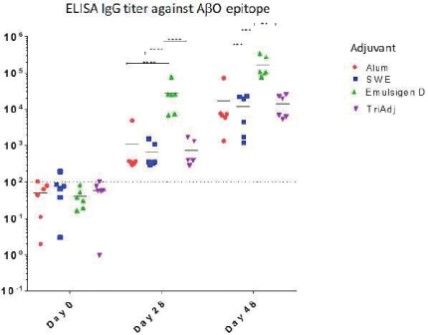
The information in this preliminary prospectus is not complete and may be changed. The securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion, dated November 1, 2022
PRELIMINARY PROSPECTUS

PROMIS NEUROSCIENCES INC.
Up to 1,383,755 Common Shares Underlying Units
Up to 345,939 Common Shares Underlying Warrants
This prospectus relates to the issuance by us and the resale by the selling security holders named in this prospectus (the “Selling Shareholders”) of up to an aggregate of 1,729,694 of our common shares, no par value (“common shares”), which consists of (i) up to 1,383,755 common shares that are issuable to certain of the Selling Shareholder that are party to the Unit Purchase Agreement, dated October 11, 2022 (the “Unit Purchase Agreement”); and (ii) up to 345,939 common shares that are issuable to certain of the Selling Shareholders that are party to the Unit Purchase Agreement upon the exercise of warrants to purchase our common shares that we issued to Selling Shareholders in a private placement that closed in connection with the Unit Purchase Agreement (the “Investor Warrants”) .
Our registration of the securities covered by this prospectus does not mean that either we or the Selling Shareholders will issue, offer or sell, as applicable, any of the securities hereby registered. The Selling Shareholders may offer, sell, or distribute all or a portion of the securities hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. We will not receive any of the proceeds from such sales of our common shares by the Selling Shareholders pursuant to this prospectus. We will, however, receive the net proceeds of any Investor Warrants exercised for cash. We will bear all costs, expenses and fees in connection with the registration of these securities, including with regard to compliance with state securities or “blue sky” laws. The Selling Shareholders will bear all commissions and discounts, if any, attributable to their sale of our common shares. See “Plan of Distribution” beginning on page 137 of this prospectus.
You should read this prospectus and any prospectus supplement or amendment carefully before you invest in our securities.
Our common shares are listed on the Nasdaq and the TSX under the symbol “PMN.” On October 28, 2022, the closing sale price of our common shares as reported on Nasdaq was $5.90 and the closing sale price of our common shares on the TSX was $8.00.
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act of 1933, as amended, and, as such, have elected to comply with certain reduced disclosure and regulatory requirements.
Our business and investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 13 of this prospectus and in the other documents that are incorporated by reference in this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is November 1, 2022.