
As confidentially
submitted to the Securities and Exchange Commission on April 4, 2022.
This draft
registration statement has not been filed publicly with the Securities and Exchange Commission, and all
information herein remains strictly confidential.
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
AMENDMENT NO. 1
To
FORM 10
GENERAL FORM FOR REGISTRATION OF SECURITIES
PURSUANT TO SECTION 12(b) OR 12(g) OF
THE SECURITIES EXCHANGE ACT OF 1934
PROMIS NEUROSCIENCES INC.
(Exact name of registrant as specified in its charter)
| Canada | 98-0647155 | |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. employer identification no.) |
Suite 200, 1920 Yonge Street
Toronto, Ontario
M4S 3E2
(Address of principal executive offices and zip code)
415-341-5783
(Registrant’s telephone number, including area code)
Securities to be registered pursuant to Section 12(b) of the Act:
Common Shares
(Title of class)
Securities to be registered pursuant to Section 12(g) of the Act:
None
(Title of class)
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | ||
| Non-accelerated filer | x | Smaller reporting company | x | ||
| Emerging growth company | x | ||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financing accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
i
Implications of Being an Emerging Growth Company
As a company with less than $1.07 billion in revenue during our most recently completed fiscal year, we qualify as an “emerging growth company” as defined in Section 2(a) of the Securities Act of 1933, as amended, which we refer to as the “Securities Act,” as modified by the Jumpstart Our Business Startups Act of 2012, or the “JOBS Act.” As an emerging growth company, we may take advantage of specified reduced disclosure and other exemptions from requirements that are otherwise applicable to public companies that are not emerging growth companies. These provisions include:
| · | Reduced disclosure about our executive compensation arrangements; |
| · | Exemptions from non-binding shareholder advisory votes on executive compensation or golden parachute arrangements; |
| · | Our election under Section 107(b) of the JOBS Act to delay adoption of new or revised accounting standards with different effective dates for public and private companies until those standards would otherwise apply to private companies; and |
| · | Exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting. |
We may take advantage of these exemptions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.07 billion in annual revenues as of the end of a fiscal year, if we are deemed to be a large-accelerated filer under the rules of the Securities and Exchange Commission (the “SEC”) or if we issue more than $1.0 billion of non-convertible debt over a three-year period.
You should rely only on the information contained in this document or to which we have referred you. We have not authorized anyone to provide you with information that is different. You should assume that the information contained in this document is accurate as of the date of this Registration Statement only.
Once this Registration Statement becomes effective (the “Effective Date”), we will become subject to the reporting requirements of Section 13(a) under Securities Exchange Act of 1934, as amended (the “Exchange Act”) and will be required to file annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and we will be required to comply with all other obligations of the Exchange Act applicable to issuers filing registration statements pursuant to Section 12(b) of the Exchange Act.
Smaller Reporting Company Status
The Company is a “smaller reporting company” as defined in Exchange Act Rule 12b-2. As a result, the Company is eligible to take advantage of certain reduced disclosure and other requirements that are otherwise applicable to public companies including, but not limited to, not being subject to the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002. The Company will remain a smaller reporting company until the last day of the fiscal year in which (1) the aggregate worldwide market value of its common stock held by non-affiliates equaled or exceeded $250 million as of the prior June 30th, or (2) its annual revenues equaled or exceeded $100 million during such completed fiscal year and the aggregate worldwide market value of its common shares held by non-affiliates equaled or exceeded $700 million as of the prior June 30th.
In this Registration Statement, unless the context otherwise requires, the terms “we,” “us,” “our,” “Company” or “ProMIS” refer to ProMIS Neurosciences Inc., together with its wholly-owned subsidiary, ProMIS Neurosciences (US) Inc.
Unless otherwise indicated, all references to “$” or “US$” in this Registration Statement refer to United States dollars, and all references to “C$” refer to Canadian dollars.
Trademarks, Trade Names and Service Marks
This Registration Statement contains certain trademarks which are protected under applicable intellectual property laws and are the Company’s property. Solely for convenience, the Company’s trademarks and trade names referred to in this Registration Statement may appear without the ® or ™ symbol, but such references are not intended to indicate, in any way, that the Company will not assert, to the fullest extent under applicable law, its rights to these trademarks and trade names.
ii
Disclosure Regarding Forward-Looking Statements
This Registration Statement contains statements that we believe are, or may be considered to be, “forward-looking statements.” Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based on current beliefs, expectations or assumptions regarding the future of the business, future plans and strategies, operational results and other future conditions of the Company. All statements other than statements of historical fact included in this Registration Statement regarding the prospects of our industry or our prospects, plans, financial position or business strategy may constitute forward-looking statements. In addition, forward-looking statements generally can be identified by the use of forward-looking words such as “plans,” “expects” or “does not expect,” “is expected,” “look forward to,” “budget,” “scheduled,” “estimates,” “forecasts,” “will continue,” “intends,” “the intent of,” “have the potential,” “anticipates,” “does not anticipate,” “believes,” “should,” “should not,” or variations of such words and phrases that indicate that certain actions, events or results “may,” “could,” “would,” “might,” or “will,” “be taken,” “occur,” or “be achieved,” or the negative of these terms or variations of them or similar terms. Furthermore, forward-looking statements may be included in various filings that we make with the SEC or press releases or oral statements made by or with the approval of one of our authorized executive officers. Although we believe that the expectations reflected in these forward-looking statements are reasonable, we cannot assure you that these expectations will prove to be correct. These forward-looking statements are subject to certain known and unknown risks and uncertainties, as well as assumptions that could cause actual results to differ materially from those reflected in these forward-looking statements.
By their very nature, forward-looking statements involve inherent risks and uncertainties, both general and specific, and risks exist that predictions, forecasts, projections and other forward-looking statements will not be achieved. We caution readers not to place undue reliance on these statements as a number of important factors could cause the actual results to differ materially from the beliefs, plans, objectives, expectations, anticipations, estimates and intentions expressed in such forward-looking statements. Risks, uncertainties and other factors which may cause the actual results, performance or achievements of the Company, as applicable, to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information and statements include, but are not limited to, the risks described under the heading “Risk Factors Summary” and in Item 1A—“Risk Factors” in this Registration Statement.
Readers are cautioned not to place undue reliance on any forward-looking statements contained in this Registration Statement, which reflect management’s opinions only as of the date hereof. Except as required by law, we undertake no obligation to revise or publicly release the results of any revision to any forward-looking statements. You are advised, however, to consult any additional disclosures we make in our reports to the SEC. All subsequent written and oral forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements contained in this Registration Statement.
iii
Investing in our securities involves risks. You should carefully consider the risks described in Item 1A—“Risk Factors” beginning on page 37 before deciding to invest in our securities. If any of these risks actually occurs, our business, financial condition and results of operations would likely be materially adversely affected. In such case, the trading price of our securities would likely decline, and you may lose all or part of your investment. Set forth below is a summary of some of the principal risks we face:
Risks Related to the COVID-19 Pandemic
| · | Our business and operations have and may be further adversely affected by the evolving and ongoing COVID-19 global pandemic. | |
| · | The ongoing COVID-19 pandemic may negatively impact the availability of scientific staff, physicians and other healthcare professionals, which may negatively impact our business, financial condition and results of operations. |
Risks Related to the Development of Our Product Candidates
| · | Our product candidates are still in the early stages of development and there is significant uncertainty that any such products will actually be developed. | |
| · | We have concentrated a portion of our research and development efforts on the treatment of AD, a field that has seen very limited success in drug development. | |
| · | Our business is heavily dependent on the successful development, regulatory approval and commercialization of PMN310 and any future product candidates that we may develop or acquire, including PMN442 and PMN267. | |
| · | Our approach to the potential treatment of AD is based on a novel therapeutic approach, which exposes us to unforeseen risks. | |
| · | We may not successfully expand our pipeline of product candidates, including by pursuing additional indications for PMN310 or by in-licensing or acquiring additional product candidates for other diseases. | |
| · | Nonclinical and clinical drug development involves a lengthy, expensive and uncertain process. The results of nonclinical studies and early clinical trials are not always predictive of future results. PMN310 or any other product candidate that we advance into clinical trials may not achieve favorable results in later clinical trials, if any, or receive marketing approval. | |
| · | Clinical failure can occur at any stage of clinical development and we have never completed a clinical trial or submitted NDA, BLA, or marketing authorization application, or MAA. | |
| · | We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates. | |
| · | Adverse side effects, properties or other safety risks associated with PMN310, PMN442, PMN267 or any future product candidates could delay or preclude approval, cause us to suspend or discontinue clinical trials, abandon further development, limit the commercial profile of an approved label or result in significant negative consequences following marketing approval, if any. | |
| · | We may experience delays or difficulties in the enrollment and retention of patients in clinical trials, which could delay or prevent our receipt of necessary regulatory approvals. | |
| · | Interim, “top-line” and preliminary results from our clinical trials that we announce or publish from time to time may change as more data become available and is subject to audit and verification procedures that could result in material changes in the final data. | |
| · | We cannot be certain that PMN310, PMN442, PMN267 or any of our future product candidates will receive regulatory approval, and without regulatory approval we will not be able to market our product candidates. | |
| · | Our lead product candidate, PMN310, is being developed for the treatment of AD, a disease that has seen limited success in drug development. | |
| · | We may in the future conduct clinical trials for our product candidates outside the U.S., and the FDA, EMA and other foreign regulatory authorities may not accept data from such trials. | |
| · | We may not be successful in our efforts to build a pipeline of additional product candidates. | |
| · | If we do not achieve our projected development goals in the time frames we announce and expect, the commercialization of our products may be delayed. | |
| · | We may develop PMN310, PMN442, PMN267 and future product candidates for use in combination with other therapies, which could expose us to additional regulatory risks. | |
| · | Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay. |
Risks Related to the Commercialization of Our Product Candidates
| · | Successful commercialization of our products, if approved, will depend on a number of factors and we cannot guarantee that we will be able to successfully commercialize our products. | |
| · | The market opportunities for PMN310, PMN442, PMN267, and future product candidates, if approved, may be smaller than we anticipate. | |
| · | Even if our current or future product candidates obtain regulatory approval, they may fail to achieve the broad degree of adoption and use by physicians, patients, hospitals, healthcare payors and others in the medical community necessary for commercial success. | |
| · | Our product candidates have never been manufactured on a commercial scale, and there are risks associated with scaling up manufacturing to commercial scale. In particular, we will need to develop a larger scale manufacturing process that is more efficient and cost-effective to commercialize our potential products, which may not be successful. | |
| · | The successful commercialization of our product candidates will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage, reimbursement levels and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those drugs and decrease our ability to generate revenue. | |
| · | We currently have no sales organization. If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our product candidates effectively in the U.S. and foreign jurisdictions, if approved, or generate product revenue. |
Risks Related to Our Financial Position and Capital Needs
| · | We will require additional financing to achieve our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our development programs, commercialization efforts or other operations. | |
| · | We have no product candidates approved for commercial sale, we have never generated any revenue from sales and we may never be profitable. |
Risks Related to Our Dependence on Third Parties
| · | We will rely on third parties to supply components, research, develop, test, and manufacture our product candidates and market, if approved. The loss of any of these third party relationships or the failure of any of them to meet their obligations to us could affect our ability to develop and obtain approval of our product candidates in a timely manner. | |
| · | We intend to rely on contract research organizations, or CROs, and other third parties to conduct, supervise and monitor a significant portion of our research and nonclinical testing and clinical trials for our product candidates, and if those third parties do not successfully carry out their contractual duties, comply with regulatory requirements or otherwise perform satisfactorily, we may not be able to obtain regulatory approval or commercialize product candidates, or such approval or commercialization may be delayed, and our business may be substantially harmed. | |
| · | If any of our third-party manufacturers encounter difficulties in production of PMN310, PMN442, PMN267 or any future product candidate we develop, or fail to meet rigorously enforced regulatory standards, our ability to provide supply of our product candidates for clinical trials or, if approved, for commercial sale could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure. | |
| · | We will likely seek collaborations with third parties for the development or commercialization of our product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of those product candidates, including PMN310, PMN442, and PMN267. |
iv
Risks Related to Our Intellectual Property
| · | If we are unable to obtain and maintain sufficient intellectual property protection for our product candidates, and other proprietary technologies we develop, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our product candidates, and other proprietary technologies if approved, may be adversely affected. | |
| · | Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time. | |
| · | If we do not obtain patent term extension for our product candidates our business may be materially harmed. | |
| · | If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business. |
| · | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. | |
| · | Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates. | |
| · | We may not be able to protect our intellectual property rights throughout the world. | |
| · | We may become subject to claims challenging the inventorship or ownership of our patents and other intellectual property. | |
| · | We may not be successful in obtaining or maintaining necessary rights to product components and processes for our development pipeline through in-licenses. | |
| · | Third-party claims alleging intellectual property infringement may prevent or delay our drug discovery and development efforts. | |
| · | We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers. | |
| · | We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming, and unsuccessful. Further, our issued patents could be found invalid or unenforceable if challenged in court, and we may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights. | |
| · | Because of the expense and uncertainty of litigation, we may not be in a position to enforce our intellectual property rights against third parties. | |
| · | If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed. | |
| · | If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected. | |
| · | Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our future products. | |
| · | Intellectual property discovered through government funded programs may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies. Compliance with such regulations may limit our exclusive rights and limit our ability to contract with non-U.S. manufacturers. |
Risks Related to Legal and Regulatory Compliance Matters
| · | Our relationships with customers, healthcare providers, including physicians, and third-party payors are subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties. | |
| · | Even if we obtain regulatory approval for PMN310, PMN442, PMN267 or any future product candidates, they will remain subject to ongoing regulatory oversight, which may result in significant additional expense. | |
| · | Failure to comply with health and data protection laws and regulations could lead to government enforcement actions and civil or criminal penalties, private litigation or adverse publicity and could negatively affect our operating results and business. | |
| · | Even if we obtain FDA or EMA approval any of our product candidates in the U.S. or European Union, we may never obtain approval for or commercialize any of them in any other jurisdiction, which would limit our ability to realize their full market potential. | |
| · | Healthcare legislative or regulatory reform measures may have a negative impact on our business and results of operations. |
| · | Our business activities may be subject to the FCPA and similar anti-bribery and anti-corruption laws. | |
| · | Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could significantly harm our business. | |
| · | Our insurance policies are expensive and only protect us from some business risks, which will leave us exposed to significant uninsured liabilities. |
Risks Related to Our Business and Industry
| · | We face significant competition in an environment of rapid technological and scientific change, and there is a possibility that our competitors may achieve regulatory approval before us or develop therapies that are safer or more effective than ours. |
Risks Related to Ownership of Our Common Shares and Our Status as a U.S. Public Company
| · | Investment in the Company’s Common Shares is speculative, involves risk, and there is no guarantee of a return. | |
| · | If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our share price and trading volume could decline. | |
| · | Concentration of ownership of our Common Shares among our existing executive officers, directors and principal shareholders may prevent new investors from influencing significant corporate decisions. | |
| · | Our constating documents permit us to issue an unlimited amount of additional Common Shares or Preferred Shares, which may prevent a third-party takeover or cause our shareholders to experience dilution in the future. | |
| · | Anti-takeover provisions in our governing documents and under Canadian Law could prevent or delay transactions that shareholders may favor. |
| · | The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation and these differences may make our Common Shares less attractive to investors. | |
| · | If we fail to attract and retain senior management and key scientific personnel, our business may be materially and adversely affected. |
| · | If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our current or future product candidates. | |
| · | We may explore strategic collaborations that may never materialize or may fail. | |
| · | We are an “emerging growth company” and a “smaller reporting company” and, as a result of the reduced disclosure and governance requirements applicable to emerging growth companies and smaller reporting companies, our Common Shares may be less attractive to investors. | |
| · | We have never paid dividends on our capital shares and we do not intend to pay dividends for the foreseeable future. Consequently, any gains from an investment in our Common Shares will likely depend on whether the price of our Common Shares increases. | |
| · | We are subject to the continued listing criteria of the TSX and our failure to satisfy these criteria may result in a delisting of our Common Shares. |
| · | We cannot assure you that our Common Shares will become listed on Nasdaq and, if listed, our failure to meet Nasdaq’s continued listing requirements could result in a delisting of our Common Shares. | |
| · | Our internal controls over financial reporting may not be effective, which could have a material and adverse effect on our business. | |
| · | The elimination of monetary liability against our directors, officers, and employees under Canadian law and the existence of indemnification rights for our obligations to our directors, officers, and employees may result in substantial expenditures by us and may discourage lawsuits against our directors, officers, and employees. | |
| · | There may be difficulty in enforcing judgments and effecting service of process on directors and officers that are not citizens of the U.S. | |
| · | If we are characterized as a passive foreign investment company, U.S. Holders may be subject to adverse U.S. federal income tax consequences. |
General Risk Factors
| · | We will incur increased costs and demands upon management as a result of being a public company. | |
| · | Comprehensive tax reform legislation could adversely affect our business and financial condition. | |
| · | Our business and operations would suffer in the event of computer system failures, cyberattacks or a deficiency in our cybersecurity or a natural disaster. | |
| · | Disruptions at the FDA, the SEC and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business. | |
| · | Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations. |
v
Glossary of Key Terms and Definitions
In this Registration Statement, unless otherwise indicated or the context otherwise requires, the following terms shall have the indicated meanings. Words importing the singular include the plural and vice versa and words importing any gender include all genders. A reference to an agreement means the agreement as it may be amended, supplemented or restated from time to time.
“Aβ” means Amyloid beta, an extracellular brain protein whose toxic misfolded form is implicated as a root cause of AD;
“AβO” means Amyloid beta oligomers; misfolded AβO are widely considered a root cause of AD;
“AChE” means Acetylcholinesterase;
“AD” means Alzheimer’s disease;
“aducanumab” means Biogen’s monoclonal antibody targeting amyloid beta;
“ALS” means amyotrophic lateral sclerosis;
“a-syn” means alpha-synuclein;
“Biogen” means Biogen Inc.;
“Board” means the board of directors of the Company;
“CBCA” means the Canada Business Corporations Act, R.S.C. 1985, c. C-44, and the regulations made under that enactment, as amended;
“CBD” means corticobasal degeneration;
“CEO” means Chief Executive Officer;
“CFO” means Chief Financial Officer;
“CMMS” means Centers for Medicare and Medicaid Services;
vi
“Collective Coordinates” means an algorithmic method which employs protein molecular dynamics to predict novel therapeutic targets in AD and other neurodegenerative diseases; and is complementary to ProMIS’ predictive algorithm technology;
“Common Shares” means the common shares in the capital of the Company;
“Company” or “ProMIS” means ProMIS Neurosciences Inc., incorporated pursuant to the CBCA on January 23, 2004 under number 4203801;
“CPT” means the Current Procedural Terminology code;
“CSO” means Chief Scientific Officer;
“DLB” means Dementia with Lewy bodies;
“DSEs” means disease specific epitopes on the molecular surface of misfolded proteins;
“DSU” refers to a deferred share unit awarded under the DSU Plan;
“DSU Plan” means the deferred share unit plan for non-employee directors;
“EMA” means the European Medicines Agency;
“FDA” means the U.S. Food and Drug Administration;
“FTLD” means frontotemporal lobar degeneration;
“GMP” means Good Manufacturing Practices;
“HD” means Huntington’s disease;
“LATE” means limbic-predominant age-related TDP-43 encephalopathy;
“LBD” means Lewy body dementia, a severe form of Parkinson’s disease;
“mAb” means monoclonal antibody;
“management” means all members of the Board as well as the senior executive officers of ProMIS;
“MCI” means mild cognitive impairment;
“MSA” means Multiple System Atrophy;
“Nasdaq” means the Nasdaq Stock Market;
“PD” means Parkinson’s disease;
vii
“PMN310” means a mAb targeting toxic prion-like forms of AβO, and designated as the Company’s first lead product candidate for development in AD;
“PMN442” means a mAb targeting toxic prion-like forms of a-syn and designated as the Company’s second lead product candidate for development in MSA;
“PMN267” means a mAb targeting toxic prion-like forms of TDP-43 and designated as the Company’s third lead product candidate;
“Preferred Shares” means preferred shares in the authorized capital of the Company;
“ProMIS USA” means ProMIS Neurosciences (US), Inc., a subsidiary corporation of ProMIS, which subsidiary was incorporated on January 14, 2016 pursuant to the General Corporation Law of the State of Delaware;
“PSP” means progressive supranuclear palsy;
“RACK1” means receptor of activated protein C kinase 1;
“Stock Option” means option granted under the terms of the Company’s Stock Option Plan;
“Stock Option Plan” means the Company’s incentive stock option plan;
“SOD1” means superoxide dismustase 1;
“TDP-43” means TAR-DNA binding protein 43;
“TSX” means the Toronto Stock Exchange and any successor thereto;
“UBC” means the University of British Columbia, Vancouver, British Columbia, Canada;
“UHN” means University Health Network, Toronto;
“USPTO” means the U.S. Patent Trademark Office; and
“U.S.” and “USA” means the United States of America, its territories, any State of the United States and the District of Columbia.
viii
Name, Address and Incorporation
ProMIS Neurosciences Inc. was incorporated on January 23, 2004 under the name 4203801 Canada Inc. pursuant to the CBCA. The Company changed its name to Amorfix Life Sciences Ltd. on August 24, 2004 and to ProMIS Neurosciences Inc. effective July 8, 2015.
See “Description of the Registrant’s Securities to be Registered” for more information about our Common Shares. The Company’s Common Shares are listed on the TSX under the symbol, “PMN.” The Company’s Common Shares also trade on the OTCQB Venture Market in the U.S. under the symbol, “ARFXF.” The Company has also submitted an application for listing of its Common Shares on the Nasdaq under the symbol “PMN.”
Our head office is located at 1920 Yonge Street, Suite 200, Toronto, Ontario, Canada M4S 3E2 and our registered and records office is located at 1055 West Georgia Street, Vancouver, British Columbia, Canada V6E 4N7. Our telephone number is (416) 847-6898 and our website address is www.promisneurosciences.com. The information provided on our website is not part of this Registration Statement.
Intercorporate Relationships
ProMIS has one wholly-owned U.S. subsidiary, ProMIS Neurosciences (US) Inc. (“ProMIS USA”), which was incorporated in Delaware on January 14, 2016. ProMIS USA has had no material activities to date.
General
ProMIS is applying its patented technology platform to build a portfolio of antibody therapies, therapeutic vaccines, and other antibody-based therapies in neurodegenerative diseases and other mis-folded protein diseases, which may include AD, MSA, ALS, PD, LBD, FTLD, PSP, CBD, and schizophrenia. These diseases share a common biologic cause – misfolded versions of proteins, that otherwise perform a normal function. The misfolded versions kill neurons and produce disease. ProMIS’ technology platform is an example of the advances in drug discovery enabled by computational power, in silico discovery, and/or artificial intelligence. We believe this platform provides a potential advantage by selectively targeting the toxic misfolded proteins with therapeutics.
ProMIS’ Platform Technology
ProMIS’ scientific foundation is centered on the growing knowledge base relating to diseases characterized by the presence of abnormal, misfolded proteins. Genetic and experimental research in the neuroscience community has demonstrated that propagating, neurotoxic, misfolded proteins (also referred to as prion-like particles or toxic soluble oligomers) are fundamental drivers of multiple neurodegenerative diseases, including AD, MSA, and ALS. ProMIS’ platform technology allows for the identification of conformational epitopes that become exposed on toxic, misfolded forms of a given protein but are not present on the properly folded form of the same protein. Such DSEs can then be used to generate therapeutic antibody candidates that selectively target toxic forms of the protein without interfering with essential functions of the healthy protein.
The Company first licensed the exclusive rights to the ProMISTM target identification technology from UBC to predict novel DSEs on the molecular surface of misfolded proteins. ProMISTM is an “in silico” rational selection approach that can be applied to any protein where the normal folding structure is at least partially known. The Company also acquired a worldwide license from UBC to “Collective Coordinates”, a method to predict novel therapeutic targets in AD and other neurodegenerative diseases. Developed principally by the Company’s former Chief Physics Officer, Steven Plotkin, PhD, the Collective Coordinates method is a computational algorithm employing protein molecular dynamics simulations, and is complementary to the ProMISTM predictive algorithm technology. The addition of Collective Coordinates to its computational discovery platform gave ProMIS a unique, proprietary and robust engine to predict DSEs on the molecular surface of misfolded proteins. The amino acid sequence of the toxic, misfolded form and the healthy, properly folded form of a target protein are exactly the same but the two differ in their shape, or conformation. The ProMIS platform offers the ability to identify targets unique to the toxic, misfolded form. These conformational epitopes are used to immunize mice or rabbits to generate selective mAbs that can attack the disease-causing form of the protein without interfering with the healthy form of the same protein. The mAbs raised in animals are humanized (the critical binding regions are inserted into a human antibody framework) for potential use in patients.
1
Our Pipeline
We are developing a pipeline of antibodies aimed at selectively targeting misfolded toxic forms of proteins that drive neurodegenerative diseases without interfering with the essential functions of the same properly folded proteins.
1 The company plans to investigate additional synucleinopathies, including PD and DLB. Arrows denote the stage of each program
2
ADDITIONAL DEVELOPMENT PROGRAMS
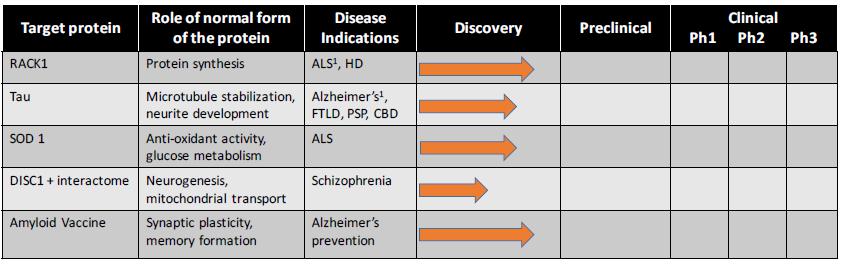
1 Initial indication
* Arrows denote the stage of each program
ProMIS’ Objectives for 2022
The Company’s priorities for 2022 fall into the following four key areas:
| · | Continue to progress PMN310 antibody lead program for AD |
| · | Continue to progress PMN442 antibody for MSA |
| · | Continue to progress PMN267 antibody for ALS |
| · | Use of the ProMIS proprietary platform to support portfolio expansion |
PRODUCT CANDIDATES
Development of a Therapy for the Treatment of AD
AD Overview
AD, a progressive neurodegenerative disease, is the most common type of dementia, accounting for approximately 60-80% of all dementia cases. Early symptoms of AD include recent memory loss, as well as apathy and depression. As the disease progresses inexorably, language deterioration, impaired ability to mentally manipulate visual information, poor judgment, confusion, restlessness, and profound mood swings develop. Eventually AD destroys cognition, personality, and the ability to function. The early symptoms of AD, especially at the inaugural stage of MCI, are often missed because they are frequently and mistakenly taken for ‘natural signs of ageing’. In 2020, current reports conclude that 50% of primary care physicians believe the medical profession is not prepared to meet the expected increase in demands the projected rise in AD and dementia cases will create.
During 2020, it was estimated there were 5.8 million Americans living with AD and that number is projected to rise to 14 million by 2050. In the U.S., one in three seniors dies of AD or another dementia, which kills more people than breast cancer and prostate cancer combined. AD is the sixth leading cause of death in the U.S., according to the Alzheimer’s Association. In 2020, AD and other dementias cost the U.S. $305 billion and those costs are projected to rise to $1.1 trillion by 2050. It is reported that 16 million Americans are unpaid caregivers, who in 2020 provided 18.6 billion hours of support, valued at $244 billion, to people with AD and other dementias.
Among people aged 70 with AD, 61% die before age 80, or twice the death rate (30%) of those without AD. Deaths from AD, as recorded on death certificates, have increased 146% over the last two decades. In the U.S. and Canada combined, an estimated 6.3 million people have AD currently, 9.5 million have MCI, and 42.3 million have evidence of potential pathology with no symptoms. In the U.S., AD costs the health system $305 billion, and that number is projected to grow to $1.1 trillion by 2050 if there are no advances in treatment and prevention.
3
Historically, a major challenge in AD has been diagnosis. Twenty years ago, diagnosis of AD could only be confirmed by autopsy. Two years ago, consensus guidelines were developed that established new diagnostic criteria – A/T/N. The methods used were based on sophisticated approaches to brain imaging: amyloid positron emission tomography (“PET”) scans measuring amyloid plaque as a proxy for pathology, tau PET scans measuring tau tangles as a proxy for pathology, and cortical magnetic resonance imaging (“MRI”) measuring the thickness of the frontal cortex of the brain as a measure of neurodegeneration. Each of these tests costs thousands of dollars, affordable perhaps to diagnose patients for a clinical trial, but not practical for screening millions of people who might be at risk or have pre-symptomatic AD.
There are now blood-based biomarkers (diagnostic assays) that can provide information that correlates with expensive A/T/N imaging so that it is possible to detect and monitor AD neuropathology in blood. Two measures have been scientifically validated: NfL, which measures the rate of neuronal loss, and phosphorylated tau which measures the level of toxic, misfolded tau. Blood levels of these have been shown to correlate with brain imaging measures, as well as disease status or progression. These advances have implications for ProMIS’ strategy. Better diagnostics can facilitate more efficient clinical trials, both in terms of identifying potential subjects for the trial and also detecting a potential treatment effect in early, small trials. Secondly, the ability to diagnose disease prior to symptoms raises the possibility of preventive treatment.
According to the World Alzheimer Report 2015, the current dementia market comprises two product categories, namely, AChE inhibitors and N-methyl-D-aspartate receptor antagonists. AChE inhibitors dominate the market. The overall market is dominated by four leading brands – Aricept, Namenda, Exelon and Ebixa. Aricept, whose active ingredient is an AChE inhibitor, holds the largest market share. The U.S. was the largest market for AD drugs in 2019, accounting for approximately 35% of total worldwide AD pharmaceutical sales in that year.
Although there is no current scientific or general consensus on the causation of AD or method of action to treat AD, evidence from some genetic and preclinical studies suggests a causative role for Ab in the pathogenesis of AD. We believe results of published genetic studies support a direct link between increased levels of Ab and disease susceptibility. Research suggests that genetic mutations in the Ab precursor protein (APP) and in the presenilin 1 and 2 genes responsible for familial forms of early onset AD all result in increased production of Ab and Ab aggregates (Citron et al, 1992; Borchelt et al, 1996). Some Down Syndrome patients with three copies of the APP gene on chromosome 21 also have elevated levels of APP and Ab deposits and have developed AD at a premature age (Podlisny et al, 1987). Along the same lines, the APOE4 allele which has been linked to an increased risk of late onset AD is associated with increased Ab deposit while the APOE2 allele linked to a decreased risk is associated with decreased Ab levels (Holtzman et al, 2012). Finally, the only known protective mutation against AD is found in the APP gene and research suggests that this leads to a reduction in the formation of Ab (Jonsson et al, 2012). In a preclinical study, it was reported that intracerebral injection of Ab-containing brain extracts from human AD patients into susceptible mice induced cerebral amyloidosis and associated pathology. Depletion of Ab from the extracts reversed this activity supporting a link between Ab and disease induction (Meyer-Luehmann et al, 2006).
While the presence of Ab plaque is a distinguishing feature of AD, there is a growing body of scientific evidence that the synaptic loss and neurodegenerative spread of AD is primarily mediated by soluble oligomers of misfolded Ab rather than plaque (Cleary et al, 2004; Jin et al, 2011). Reports from several groups indicate that plaque burden correlates poorly with memory impairment (Cleary et al, 2004; Ferreira et al, 2015) and insoluble Ab fibrils show little or no demonstrable toxicity in vitro or in vivo (Balducci et al, 2010; Shankar et al, 2008). In contrast, a significant correlation between disease severity and levels of soluble Ab in the central nervous system was reported by Lue et al (Lue et al, 1999), and the direct neurotoxicity of soluble Ab oligomers was demonstrated in neuronal cultures in vitro by separate groups (Lauren et al, 2009; Jin et al, 2011). In published reports using rodent models, the injection of soluble oligomeric Ab, but not soluble monomers or plaque, was shown to induce synaptic damage and cognitive dysfunction (Cleary et al, 2005; Hong et al, 2016).
4
A convergence of evidence from multiple studies suggests that the progressive nature of AD arises from the formation and spread of a prion-like subset of misfolded oligomers of Ab that adopt a b-sheet-rich conformation transmissible to native Ab in a template-like manner. The self-propagation of these prion-like oligomers follows the stereotypical progression of AD with initial involvement of the enthorhinal cortex followed by spreading to the hippocampus and neocortex as described by Khan et al (Khan et al, 2014). The prion-like spread of Ab oligomers has been well-documented in animal models by different groups following the injection of purified oligomers or brain extracts from AD patients or diseased animals (Cleary et al, 2005; Meyer-Luehmann et al, 2006; Watts et al, 2014; Hong et al, 2016). There is also in vitro evidence that such misfolded “Ab prions” from AD brain can catalyze the misfolding and hyperphosphorylation of tau, another protein involved in the pathogenesis of AD as reported by Jin et al (Jin et al, 2011). Targeting of Ab oligomers therefore represents an attractive strategy to inhibit progression of the neurodegenerative Ab-Tau cascade (Choi et al, 2015; Khan et al, 2014).
PMN310
ProMIS’ lead therapeutic program is PMN310, a mAb designed to treat AD by selectively targeting the toxic misfolded form of Aβ. Based on the understanding of Ab biology described above, PMN310 was designed to be more selective for the toxic oligomer of amyloid than aducanumab, Biogen’s anti-Aβ antibody, and BAN 2401, currently being co-developed by Eisai Co. and Biogen. Both aducanumab and BAN2401 bind oligomers, but also plaque. This off-target binding of plaque frequently leads to a side effect, ARIA-E, and potentially limits the benefit of aducanumab and BAN2401 by both limiting the highest dose that can be safely administered and by “wasting” a substantial portion of the administered antibody which binds plaque, reducing what is available to neutralize the toxic oligomers.
Although there is no current scientific or general consensus on the causation of AD or method of action to treat AD, the purported importance of targeting toxic oligomers is supported by recent results of clinical trials, conducted by third parties, with therapeutic mAbs targeting Ab. Antibodies that bind Aβ monomers (bapineuzumab, solanezumab, crenezumab) did not show efficacy, suggesting that high selectivity for low abundance toxic AβO is desirable to prevent mAbs from being consumed by unproductive binding to non-pathogenic, abundant monomers (target distraction). Other antibodies with reduced binding to monomers and more selectivity for aggregated Aβ produced more promising results, including aducanumab which received accelerated approval from the FDA, and BAN2401 and donanemab which showed evidence of a cognitive benefit in Phase 2 trials. However, treatment with all of these antibodies was associated with the dose-limiting adverse events of ARIA-E (brain edema) and ARIA-H (microhemorrhages) correlated with binding to insoluble deposits of Ab in the vasculature and plaque. We believe that a selective, oligomer-specific antibody that does not bind monomers or plaque would circumvent these issues and potentially provide an improved product profile. We believe PMN310 displays these desired characteristics (see below).
Using the ProMIS platform, six different conformational epitopes were identified as potential targets exposed on toxic AβO but not Aβ monomers or plaque. MAbs raised against these epitopes displayed selectivity for Aβ oligomers vs monomers and inhibited AβO toxicity and propagation in vitro. In January 2017, the Company designated the PMN310 antibody (binds conformational epitope 301) as its first lead candidate for development in AD. As described in our published preclinical studies (Gibbs et. al., 2019), PMN310 displayed the desired selective profile with binding to synthetic AβO and little or no binding to Aβ monomers as determined by surface plasmon resonance (SPR), and no detectable binding to plaque or vascular deposits in AD brain sections as determined by immunohistochemistry (IHC). In SPR studies with cadaveric brain tissue from individuals with AD, PMN310 also showed binding to brain fractions indicating that PMN310 can potentially recognize toxic AβO species present in the brain. In vitro, PMN310 inhibited AβO propagation in a thioflavin-T (ThT) based assay measuring the formation of Aβ aggregates with a beta-sheet structure over time (Fig. 1). PMN310 also reduced the killing of primary mouse neurons by toxic AβO in culture (Fig.1). In vivo, the activity of murine PMN310 was tested in two different models. In one model conducted at SynAging (Vandoeuvre-les-Nancy, France), PMN310 and a preparation of toxic AβO were co-delivered (mAb:AβO ratio of 1.6) by intracerebroventricular (ICV) injection into male, 3-month old, wild-type C57Bl6/J mouse to determine whether PMN310 might improve cognitive performance and molecular markers in this model of AβO-induced neurotoxicity. Treatment groups consisted of day 0 ICV injection of vehicle alone, AβO alone, vehicle with PMN310 or AβO with PMN310, and contained 12 mice per group to achieve statistical significance. Cognitive performance was assessed on days 7-8 using the novel object recognition (NOR) assay. Mice were sacrificed and perfused on day 10, the hippocampus was isolated and levels of synaptic (PSD-95, SNAP25) and inflammation (TNF-a) markers were measured by ELISA in hippocampal homogenates from individual mice. AbO-injected mice failed to recognize a new object and displayed a discrimination index of 0 or less. Co-injection of PMN310 with the toxic oligomers prevented this cognitive deficit. As expected, ICV injection of PMN310 alone had no effect (Fig. 2). The cognitive deficit induced by ICV injection of AbO was associated with inflammation and synaptic damage in the hippocampus, a region important in the development of memory. Hippocampal homogenates from AbO-treated mice displayed an increase in levels of TNF-a and decreases in PSD-95 and SNAP25. Partial protection from these changes was observed in mice co-injected with AbO and PMN310.
5
| Figure 1A | 1B |
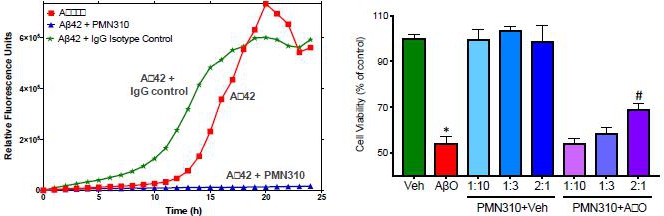
Fig. 1. Inhibition of aggregation propagation in vitro (thioflavin-based assay) (A) and inhibition of AβO toxicity for primary mouse neurons in vitro (B).
Figure 2
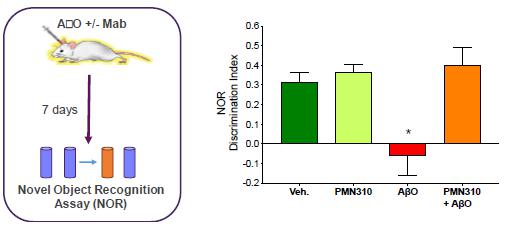
Fig. 2. Administration of PMN310 to mice prevented the loss of short-term memory formation caused by toxic AβO.
*p<0.05 vs Vehicle, #p<0.05 vs AβO. Discrimination index = (time exploring new object – time exploring familiar object)/total exploration time.
In a second in vivo model conducted at reMYND (Leuven, Belgium), the potential effect of treatment with murine PMN310 (mouse IgG2a) was tested in the transgenic (Tg) hAPP[V717I] mouse model of AD. Characterization of the model indicates that these hAPP-Tg mice display spontaneous, progressive accumulation of Ab in the brain, eventually resulting in amyloid plaques around 10-11 months of age. In the pre-plaque stage of the pathology, there is a clear cognitive and long-term synaptic potentiation (LTP) deficit in these mice suggesting that impairment is caused by soluble toxic species such as AbO rather than plaque. The aim of the study was to assess the impact of 7 weekly doses of PMN310 administered intraperitoneally (i.p.) at 30 mg/kg to female mice, beginning at 5.0 months of age. Experimental groups consisted of hAPP-Tg mice treated with vehicle or PMN310, and non-Tg, age-matched littermates treated with vehicle as a control, with 17 mice per group to achieve statistical significance. Spatial learning and memory performance were assessed using the Morris Water Maze task at 6.4 months of age (after 7 doses of antibody) which measures the ability of mice to learn and remember the location of a hidden platform in a pool of water. Compared to non-Tg littermates, the hAPP-Tg mice were significantly impaired and showed an increase in both escape latency (time required to find the hidden platform, p=0.0024) and the search path or distance traveled to reach the platform (p=0.0047). Treatment of hAPP-Tg mice with PMN310 significantly improved these outcomes with a decrease in escape latency (p=0.0187) and search path (p=0.0071) (Fig. 3).
6
Figure 3
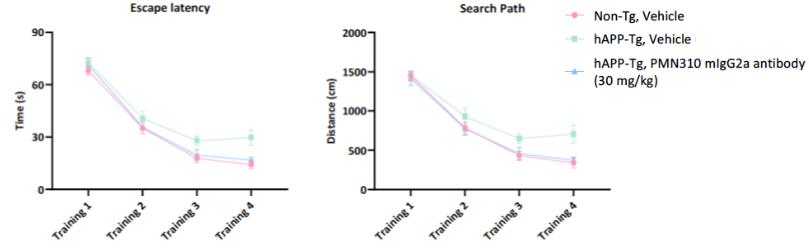
Fig. 3. Systemic administration of PMN310 provides a cognitive benefit in a mouse model of AD (hAPP[V717I] Tg mice)
As described in our publication (Gibbs et al, 2019), PMN310 brain exposure and kinetics after systemic i.p. administration were assessed in mice. In one study conducted by ProMIS, aged 15-17 month old wild type littermates of APP/PS1 mice, received a single 30 mg/kg i.p. injection of humanized PMN310 (n=4), aducanumab (n=3) or PBS as a negative control (n=2). Levels of human IgG present in the plasma and perfused brains were measured 24 h later by ELISA. Equivalent amounts of PMN310 and aducanumab were detected in plasma and brain demonstrating a comparable degree of CNS penetrance (p=0.28) in the range of ~0.3%. As expected, no human IgG was detected in mice injected with PBS alone. Additionally, a study was conducted by ProMIS in aged (13-17 months old) transgenic APP/PS1 mice in order to assess the time course of CNS exposure to PMN310. Plasma and brain levels of human IgG were measured by ELISA on days 1, 7, 14 and 21 after i.p. administration of 30 mg/kg PMN310 (n=4-6 per time point). In spite of declining plasma levels (p=0.0016 for day 1 vs day 7, p<0.0001 for day 1 vs days 14 and 21), CNS levels of PMN310 were detectable out to the study endpoint at day 21 (no significant difference in brain levels at the different time points). These results suggest that PMN310 is comparable to other therapeutic mAbs and is similarly able to cross the blood-brain barrier to reach its target. The Company believes that the greater selectivity of PMN310 for AβO may result in greater neutralization of this disease-causing species (no target distraction) and may potentially be better tolerated (allowing for higher doses) due to a reduced risk of the ARIA adverse events that have been reported associated with plaque-binding antibodies.
7
The Company successfully humanized PMN310 in a human IgG1 framework. Producer cell line development is progressing using Selexis' proprietary and high performance SUREtechnology Platform. Cell banks and drug product candidate will be generated in partnership with KBI. An Investigational New Drug Application (“IND”) for Phase 1 testing of PMN310 is planned for 2022.
Development of a Therapy for the Treatment of MSA
MSA Overview
MSA is a rare neurodegenerative disease with an estimated prevalence of 3.4-4.9 cases per 100,000 population. MSA is characterized by rapidly progressive autonomic failure and motor symptoms with predominant parkinsonian features (MSA-P) or dominant cerebellar features (MSA-C). There is no effective treatment and the mean survival from the onset of symptoms is 6-10 years. Histologically, the disease is characterized by a-syn aggregates in the cytoplasm of oligodendrocytes and, to a lesser extent, in neurons and other glial cells. The causative role of a-syn aggregates in MSA pathogenesis is supported by experimental evidence showing that a-syn aggregates from MSA brain homogenates propagate in a prion-like manner in vitro and in vivo, and cause MSA-like neurodegeneration in mice. The characteristics of MSA, although devastating for the patients, present several advantages for clinical testing: disease progression is rapid allowing for earlier detection of therapeutic potential; high levels of NfL in serum represent a potential biomarker for inhibition of neuronal damage; and no placebo effects have been observed in clinical trials to date. Even though MSA is a rare disease, recruitment is facilitated by the unmet need and existence of a global MSA Registry (GLOMAR), along with supporting organizations.
8
PMN442
Research discussed in the literature indicates that misfolded toxic a-syn is a primary driver of disease. Multiple studies indicate that pathogenic aggregates of a-syn can propagate from cell to cell in a prion-like manner causing progressive neuronal damage and disease symptoms. In order to target pathogenic a-syn without interfering with normal a-syn, the ProMIS platform was used to generate several mAbs against predicted conformational epitopes of misfolded, toxic a-syn. These mAbs showed the ability to selectively bind the pathogenic forms of a-syn (toxic oligomers and small soluble fibrils) but not the normal forms of a-syn (monomers, physiologic tetramers) that play important functional roles in the brain (Fig. 4). In vitro studies showed that the mAbs were able to protect rat dopaminergic neurons (neurons that are destroyed in PD) against toxic a-syn oligomers (Fig. 5). In separate assays, the mAbs also neutralized the seeding activity of a-syn soluble fibrils (Fig. 6) which is involved in the cell to cell propagation of disease. Importantly, the mAbs recognize toxic species of a-syn present in brain homogenates from patients with synucleinopathies (Fig. 7). Taken together, the results support the potential for using these antibodies to selectively target and protect against a-syn pathogenic species in patients with synucleinopathies.
Figure 4
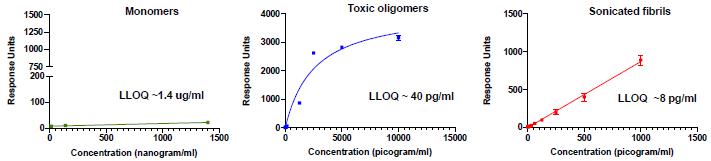
Fig. 4. Selectivity of mAbs for pathogenic species of a-syn. The binding response of a representative mAb to various concentrations of a-syn monomers, toxic oligomers and soluble fibrils (sonicated PFFs) measured in a Millipore immunoassay. Mean + SD of triplicates shown with the calculated lower limit of quantitation (LLOQ) for each species.
Figure 5
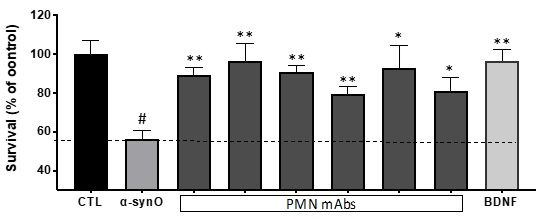
Fig. 5. Protection against neurotoxicity. mAb inhibition of oligomer toxicity for dopaminergic neurons. Cultures of primary rat dopaminergic neurons were exposed to toxic a-syn oligomers with or without mAbs. Survival is expressed as the percentage of viable neurons compared to a control culture with vehicle only (CTL). Results shown are the mean + SEM of 6 replicate cultures. BDNF was used as a positive control. # p = 0.0004 vs. CTL, *p < 0.002 vs. a-synO, **p < 0.003 vs. a-synO.
9
Figure 6
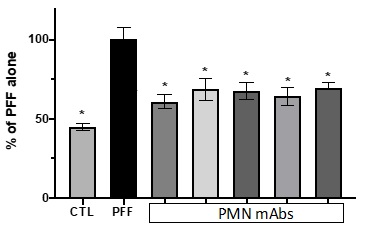
Fig. 6. Inhibition of seeding activity. mAb inhibition of the recruitment of endogenous rat a-syn into phosphorylated aggregates. Cultures of primary rat hippocampal neurons were exposed to soluble human a-syn preformed fibrils (PFF) with or without mAbs. CTL = neurons incubated with vehicle alone. Results are expressed as a percentage of the phosphorylated rat a-syn staining area with PFF alone and show the mean + SEM of 6 replicate cultures. *p<0.02 vs PFF.
Figure 7
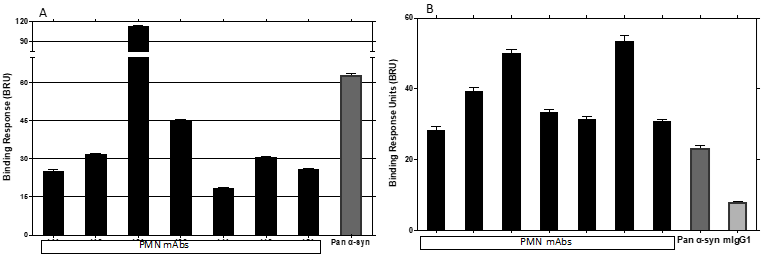
Fig.7. Binding to native pathogenic a-syn species in patient brain extract. The binding response of immobilized mAbs to a-syn in brain extract from DLB (A) and MSA (B) patients was measured by SPR. A pan a-syn reactive antibody and mouse IgG1 (mIgG1) were used as controls. Results shown are the mean + SEM of two (A) or four (B) independent studies.
Using the ProMIS platform, several conformational epitopes were identified as likely to become exposed on misfolded, pathogenic forms of a-syn (toxic oligomers and soluble seeding fibrils). MAbs were raised against these epitopes and were tested for the desired binding profile and ability to protect neurons against toxic a-syn species in vitro. Multiple mAbs were screened and PMN442 emerged as the lead candidate for this program with the desired characteristics. PMN442 showed robust binding to a-syn oligomers and seeding fibrils, with negligible binding to a-syn monomers and physiologic tetramers which are required for normal neuronal function. PMN442 also reacted with native toxic a-syn present in brain homogenates from individuals with MSA and DLB. In addition, PMN442 did not stain dense deposits of a-syn in Lewy bodies and Lewy dendrites in brain sections from diseased individuals. Although characteristic of disease, Lewy bodies/Lewy dendrites are not believed to be a major driver of toxicity and can actually act as a sink, diverting antibodies away from the pathogenic species (oligomers and seeding fibrils).
10
In activity assays, PMN442 protected rat dopaminergic neurons against killing by a-syn toxic oligomers. PMN442 also inhibited the processes involved in the cell-to-cell propagation of a-syn aggregates: it reduced the uptake of human a-syn seeding fibrils by neurons and the subsequent formation of intracellular aggregates, as well as the recruitment of endogenous normal a-syn into those aggregates. These results support the potential of PMN442 to selectively target and protect against a-syn pathogenic species in patients with MSA and other synucleinopathies.
Development of a Therapy for the Treatment of ALS
ALS Overview
ALS, commonly known as Lou Gehrig’s Disease, is a progressive neurodegenerative disease that causes muscle weakness, paralysis and, ultimately, respiratory failure. ALS attacks randomly, occurs throughout the world with no racial, ethnic or socioeconomic boundaries. Most people with ALS only live two to five years after their first signs of disease. It is estimated there are currently 30,000 people in North America and 450,000 people worldwide, suffering from ALS, with approximately 5,000 new cases arising in North America annually. Patients with ALS present symptoms such as progressive weakness, muscle atrophy and spasticity. These neurodegenerative and neuromuscular symptoms arise due to the ultimate degeneration of motor neurons in the spinal cord, the brain stem and in the brain cortex. Incurable and usually fatal within five years, ALS gradually robs a patient of the ability to walk, talk and breathe. Currently, there is no confirmatory test for ALS and many people go undiagnosed at early phases of the disease. Approximately two-thirds of those afflicted by ALS are currently undergoing some form of symptomatic treatment. There are no therapies approved that halt or significantly slow progression.
The biological mechanisms that cause ALS are only partially understood. There are scientific research results indicating that toxic, misfolded forms of TDP-43 are believed to play an important role in the development of ALS.
PMN267
Misfolded, aggregated TDP-43 forming inside neurons has been implicated in the pathogenesis of ALS, FTLD and LATE through direct toxicity, loss of function of normal TDP-43, induction of misfolding of other neuronal proteins, and prion-like, cell-to-cell propagation of disease.
Experimentally, misfolded aggregates of TDP-43 are toxic to neural cells, and the prion-like propagation of TDP-43 aggregates has been demonstrated in cell culture and animal models. Importantly, misfolded TDP-43 has been found to induce the misfolding of other proteins into pathogenic aggregates (e.g., SOD1, nuclear pore proteins and transport proteins, DISC1), such that targeting misfolded TDP-43 potentially represents an opportunity to not only neutralize TDP-43 pathology but also interrupt this pathogenic interactome.
Identification of epitopes present on misfolded TDP-43 through the ProMIS discovery platform allowed for the generation of high affinity antibodies (Fig. 8) showing selective recognition of misfolded cytoplasmic aggregates of TDP-43 with no detectable interaction with normal TDP-43 which is located in the nucleus and is important for normal cell function (Fig. 9). The antibodies also recognized and stained pathogenic TDP-43 aggregates in spinal cord sections from ALS patients and brain sections from FTLD patients (immunohistochemistry) indicating that they have the potential to target disease-causing TDP-43 in these patients. In vitro data showed that such antibodies can inhibit the cell to cell transmission of misfolded TDP-43 in the extracellular space thereby offering the potential to inhibit spreading of pathology (Fig. 10).
11
Figure 8
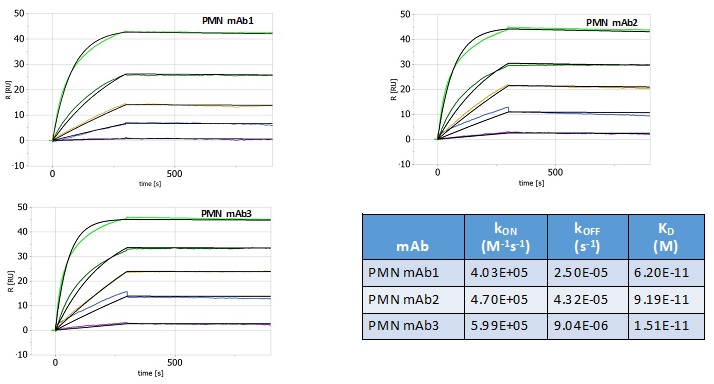
Fig. 8. High affinity mAbs. In SPR studies, serial dilutions of test mAbs were flowed over the target epitope immobilized on sensorchips to assess the binding kinetics and affinity. Binding curves were fitted to a Langmuir 1:1 interaction model.
Figure 9
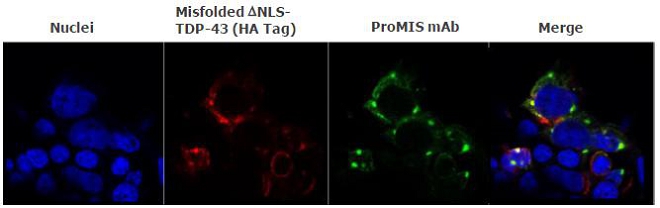
Fig. 9. Selective binding of mAb to misfolded, cytoplasmic aggregates of TDP-43. Staining of HEK293 cells transfected with mutant TDP-43 shows cytoplasmic aggregates of misfolded TDP-43 (red). Staining of the same cells with a PMN mAb (green) shows co-localization with TDP-43 aggregates with no staining of endogenous, normal TDP-43 in the nucleus (nuclei stained blue).
12
Figure 10
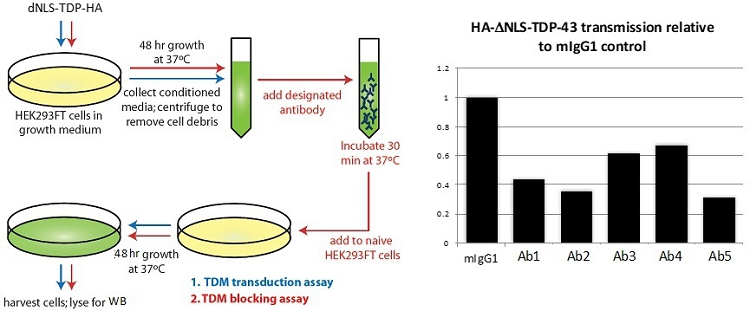
Fig. 10. Inhibition of cell-to-cell transmission of misfolded TDP-43 by mAbs. Supernatant from HEK293 cells transfected with misfolding mutant TDP-43 was incubated with test antibodies and added to naïve recipient cells to assess transmission of misfolding TDP-43 (HA-tagged). Compared to a mouse IgG1 negative control (mIgG1), several mAbs inhibited transmission to recipient cells as determined by a reduction in the density of the HA band on a Western blot of recipient cell lysate.
Intrabody versions of the TDP-43 antibodies were also generated. Intrabodies (from intracellular and antibody) are expressed from within the cell and were designed to target intracellular aggregates of TDP-43. Testing indicated that intrabodies expressed inside HEK293 cells associated selectively with pathogenic aggregates of TDP-43 in the cytoplasm (Fig. 11) and promoted degradation of the aggregates without affecting normal TDP-43 function or harming the cells (Fig. 12).
Figure 11
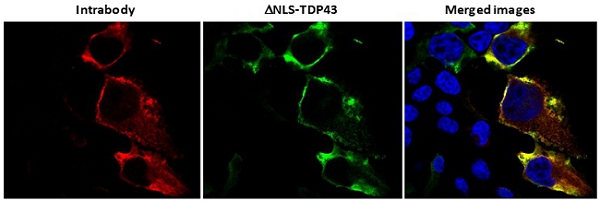
Fig. 11. Co-localization of intrabody with misfolded, cytoplasmic aggregates of TDP-43. Staining of HEK293 cells co-transfected with mutant TDP-43 (green) and plasmid encoding a PMN intrabody (red) shows co-localization of the two. There was no interaction of the intrabody with endogenous, normal TDP-43 in the nucleus (nuclei stained blue).
13
Figure 12
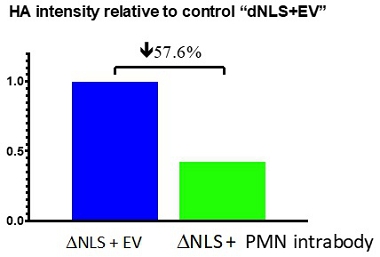
Fig. 12. Clearance of TDP-43 aggregates by intrabody. Transfection of HEK293 cells with a ProMIS intrabody results in degradation of HA-tagged mutant TDP-43 (dNLS) aggregates as measured by reduction in the density of the HA band on a Western blot of cell lysate compared to an empty vector (EV) control.
These results support the potential for using these mAbs to selectively target and protect against pathogenic TDP-43. We believe the antibodies could be used to interfere with the cell to cell spread of misfolded aggregates of TDP-43 in the extracellular space while intrabody constructs delivered inside the cells via gene therapy vectors could be used to degrade intracellular aggregates and prevent further propagation (concept illustrated in Fig. 13).
Figure 13

Using the ProMIS platform several epitopes were identified as likely to become exposed on misfolded, pathogenic forms of TDP-43, but not on the properly folded, functional protein. MAbs were raised against these epitopes and tested for selective reactivity with misfolded TDP-43 aggregates and protective activity. Screening of multiple mAbs yielded PMN267 as the lead candidate exhibiting the desired properties. PMN267 bound its target epitope with high affinity in the 10E-11M range. In a cell system, PMN267 showed selective recognition of misfolded, cytoplasmic TDP-43 aggregates and no detectable interaction with endogenous normal TDP-43 in the nucleus. Similarly, PMN267 did not react with TDP-43 in stress granules which are important in protection against oxidative stress. PMN267 also showed binding to exosomes derived from the brains of deceased FTLD individuals. In vivo studies are ongoing.
An intrabody version of PMN267 (single chain antibody sequence encoded into a plasmid) expressed from within cells showed co-localization with cytoplasmic aggregates of TDP-43 and no detectable binding to normal, nuclear TDP-43. Expression of the intrabody promoted degradation of the misfolded TDP-43 aggregates. The Company believes that the observed selectivity of PMN267 for misfolded TDP-43 and avoidance of normal TDP-43 has the potential to allow for inhibition of disease without compromising essential TDP-43 function. Intrabody development would involve collaboration with a partner with gene therapy expertise.
14
DEVELOPMENT PROGRAMS
Expansion to Include Other Neurodegenerative and Mis-folded Protein Diseases
The ProMIS discovery platform is also being applied to other toxic proteins that drive neurodegenerative and other mis-folded protein diseases including a-syn in PD and LBD, tau in AD, FTLD, PSP, and CBD, RACK1 in ALS and HD, SOD1 in ALS and DISC1 in schizophrenia. Under disease conditions, misfolding of these proteins leads to the formation of toxic aggregates inside brain cells that are capable of spreading damage by propagating from cell-to-cell. Disease-associated conformational epitopes identified through ProMIS’ computational platform can be used to potentially generate therapeutic antibodies or potentially form the basis for the development of vaccines. The Discovery phase of the process comprises 2 distinct stages: 1) computational modeling to predict and construct conformational peptide epitopes present on the misfolded, toxic form of a protein, followed by either immunization with the peptide epitopes to generate antibodies/intrabodies, or incorporation of the peptide antigen into a therapeutic vaccine, 2) screening and validation of multiple candidates in vitro and in vivo to select a lead for preclinical development.
Alpha-synuclein
Evidence from genomic analysis, cell culture, and in vivo studies points to a-syn as a major driver of PD and other synucleinopathies. A large body of data suggests that soluble aggregates (oligomers, soluble fibrils) are the most toxic form of a-syn and can spread pathology from neuron to neuron. In contrast, a-syn monomers and physiological tetramers are non-toxic and are required for normal neuronal function.
In an attempt to develop a potential therapy, the ProMIS computational platform was used to identify epitopes that are selectively exposed on toxic misfolded species of a-syn, in a conformation that distinguishes it from that of monomers, physiological tetramers and insoluble fibrils (Lewy body/Lewy neurites). The analyses suggest that immunization with these conformational epitopes led to the generation of mAbs that selectively bind the toxic forms of a-syn as determined by several methods (surface plasmon resonance, immunohistochemistry and dot blot analysis).
PD is a progressive neurodegenerative disorder characterized by loss of dopaminergic neurons located in the midbrain and the presence of intraneuronal inclusions (Lewy bodies/Lewy neurites) consisting mainly of aggregates of a-syn. Accumulation of insoluble a-syn fibrils in the brain is also observed in LBD.
PD is the second most common neurodegenerative disorder after AD. It is estimated there are over 10 million patients diagnosed worldwide and up to 10 million total subjects with PD, including undiagnosed individuals. The prevalence of PD increases with age, affecting approximately 1.1% of the population over the age of 60. Currently, the Parkinson’s Foundation Prevalence Project estimates 930,000 people in the U.S. live with PD, and this number is expected to rise to 1.2 million by 2030. As global life expectancy increases, so will the burden of PD. It is estimated that the global number of people with PD will grow by 88% between 2020 and 2040.
Global Data estimates that drug sales for PD will, according to the U.S. National Institute of Environmental Health Sciences, grow from 2020 to 2027 by approximately 11.3% to reach $4.76 billion across the seven largest markets by 2027. Over the seven year forecast period, the market is expected to grow driven by the launch of late-stage pipeline products and growth of the PD population. North America dominates the PD market. Nearly 1 million people in the U.S. are living with PD with the average age of onset being 60 and the prevalence of PD in the population increases from 1% at age 60 (5 – 10% of whom had early onset from age 50) to 4% at age 80. It is predicted by the U.S. National Institute of Health that by 2030, due to the aging population in the U.S., approximately 1.2 million people will be suffering from PD. The Asia Pacific market is also expected to record significant growth in this market, centered on China, Japan and India.
15
Scientific studies indicate that toxic oligomers and small soluble fibrils, derived from naturally occurring a-syn, are the root cause of disease development and progression in PD. Recent findings suggest that physiological a-syn tetramers inhibit aggregation and must be preserved for normal a-syn homeostasis. We believe a potential therapy will require antibodies selective for the toxic forms of a-syn oligomers and/or small soluble fibrils, while avoiding physiologic forms of a-syn. Selectivity for only the toxic forms of a-syn represents the essential feature of a successful antibody therapy, for it is critical that treatment not hinder normal forms of a-syn that play an important functional role in the brain. Traditional methods are unable to generate antibodies with adequate precision to selectively target these neurotoxic forms of a-syn. ProMIS is using its proprietary technology platform for discovering and developing antibodies that can uniquely and precisely target these specific toxic forms.
Current treatments center on the management of dopamine levels in the brain, with levodopa-based therapies remaining the standard of care in the PD market for the past 50 years. These therapies provide initial relief of symptoms at a low annual cost of therapy, with Sinemet (carbidopa/levodopa, Merck Sharp & Dohme) and Madopar (benserazide/levodopa, Roche) being the two most popular branded drugs. However, dopamine replacement or dopamine agonist (bromocriptine) therapies only provide temporary symptomatic relief, are associated with debilitating long-term side effects and do not affect the underlying neurodegeneration and progression of disease related to toxic forms of a-syn.
Tau
Propagation of misfolded, pathogenic aggregates of tau has also been implicated in AD and other tauopathies such as PSP, CBD, and FTLD-tau. The ProMIS platform was therefore used to identify epitopes and raise mAbs against pathogenic forms of tau (toxic oligomers and small soluble fibrils). A set of mAbs has been generated that preferentially bind pathogenic tau aggregates as opposed to physiologic tau monomers. The Company believes that selectivity of antibodies for tau pathogenic species, as opposed to pan-tau reactivity (binding to all forms of tau), is needed both in order to preserve normal tau function and to minimize the diversion of active antibody from the target through unproductive binding to more abundant non-toxic forms of tau. In binding assays, the ProMIS mAbs recognized toxic species of tau in brain homogenates from individuals with AD (Fig. 14) indicating that the mAbs can recognize toxic tau species present in the brain. In activity assays, the mAbs were able to inhibit the seeding activity of AD brain homogenate resulting in decreased induction of tau aggregation in a cell system. (Fig. 15). These results suggest that these mAbs may be useful in targeting pathogenic tau in AD and potentially other tauopathies.
Currently, mAbs against additional epitopes are being generated and screened. The next steps will entail selecting the candidates for in vivo testing and eventual selection of additional product candidates.
16
Figure 14
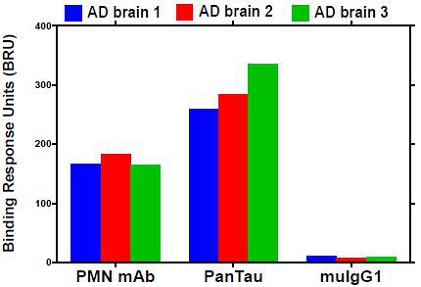
Fig. 14. Binding to native pathogenic tau species in the brain extracts of individuals with AD. The binding response of a representative immobilized mAb to tau in brain extract from 3 different individuals with AD was measured by SPR. Mouse IgG1 (muIgG1) was used as a negative control.
Figure 15
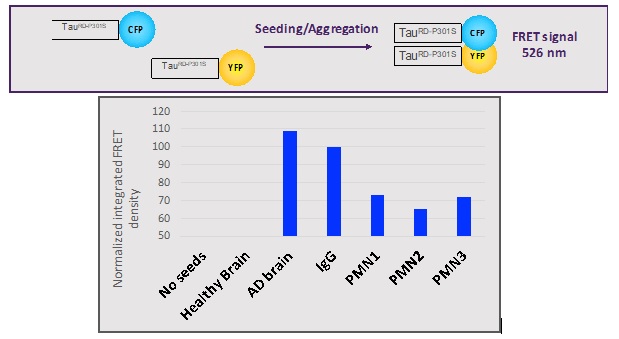
Fig. 15. Inhibition of seeding activity of AD brain homogenate. Brain homogenate +/- mAbs was transduced into Biosensor cells with Lipofectamine 200. FRET signal was measured 48 hours later by flow cytometry. Results are expressed as Normalized Integrated FRET density defined as the percent of FRET positive cells multiplied by the Median Fluorescence Intensity of those FRET positive cells and normalized to cells treated with IgG.
The immediate goal for the ProMIS extended therapeutic portfolio is to achieve further scientific characterization and validation of one or more of these programs. Based on results of this evaluation the most promising program or programs will be advanced in development.
17
SOD1
Misfolded SOD1 is abundant in both familial (FALS) and sporadic (SALS) forms of ALS but is not found in samples from normal controls or patients with other neurodegenerative disorders. Misfolding of SOD1 can be induced by mutations (FALS) or other factors such as oxidative stress and interaction with misfolded TDP-43, as mentioned above. Like TDP-43, misfolded SOD1 aggregates are toxic and have the ability to propagate from cell to cell causing spreading of disease. In order to target pathogenic SOD1 in ALS, the ProMIS platform was used to successfully generate mAbs against different regions of SOD1 predicted to be exposed in misfolded SOD1 but inaccessible in the normal, properly folded protein. These antibodies were shown to bind misfolded SOD1 in spinal cord homogenates from both FALS and SALS patients, with no recognition of normal SOD1 in control normal spinal cord.
In in vitro assays, ProMIS mAbs inhibited the cell to cell propagation of misfolded SOD1 while administration of mAb in a mouse model of ALS significantly prolonged survival of the animals. These results support the potential use of such mAbs to selectively target and protect against pathogenic SOD1 in ALS.
Currently, mAbs against additional epitopes are being generated for screening. The next steps will entail additional in vitro and in vivo studies for eventual selection of a lead(s) for development.
RACK1
RACK1 is a core ribosomal protein of the eukaryotic small (40S) ribosomal subunit. It is a scaffold protein that interacts with several other proteins thereby regulating a variety of signaling pathways critical for cell proliferation, transcription and protein synthesis. It is essential for proper neuronal function. In ALS, our own findings and those of others indicate that misfolded RACK1 co-localizes in cytoplasmic aggregates in motor neurons of the spinal cord suggesting an involvement in disease. Indeed, in a cell system, mutant TDP-43 has been reported to suppress global protein synthesis by co-aggregating with RACK1 on polyribosomes, a finding that we have reproduced.
To investigate RACK1 as a potential target for ALS and HD, ProMIS explored the impact of RACK1 knock-down (KD) in vitro and in vivo (i.e., what happens in the absence of RACK1). In a cell system, as expected, RACK1 was observed to co-aggregate with misfolded mutant TDP-43 in the cytoplasm. Knock-down of RACK1 expression resulted in disaggregation of cytoplasmic TDP-43 and even relocation to the nucleus (normal location) in some of the cells (Fig. 16). The disaggregation was accompanied by a reversal of the suppression of protein synthesis by mutant TDP-43 (Fig. 17).
18
Figure 16

Fig. 16. Disaggregation of mutant TDP-43. RACK1 (red) normally co-localizes with mutant TDP-43 aggregates in the cytoplasm (green) in the top panels. With RACK1 KD (bottom panels), TDP-43 disaggregates and localizes in the nucleus in some of the cells.
19
Figure 17
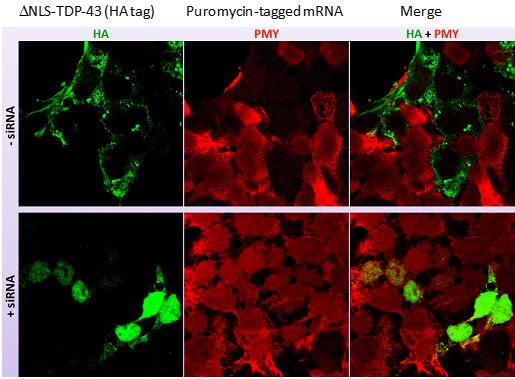
Fig. 17. Reversal of protein synthesis suppression. Protein synthesis (red) is suppressed in cells expressing aggregated mutant TDP-43 (green) in the top panels. RACK1 KD (bottom panels) disaggregates TDP-43 and restores protein synthesis.
In fruit fly studies, genetic KD of RACK1 in retinal neurons protected against neurodegeneration caused by overexpression of TDP-43 (Fig. 18)

Fig. 18. Protection against neuronal degeneration. Fruit flies overexpressing human TDP-43 in retinal neurons show degeneration (loss of ommatidia pointed by arrow). TDP-43 pathology is prevented by RACK1 KD in these flies.
Results from the literature and ProMIS’ proof of concept data using RACK1 KD support intracellular targeting of RACK1 as a potential therapeutic approach for ALS and HD. Knock-down of RACK1 is non-selective (all forms knocked-down) and may not be well-tolerated in humans. We are therefore using the ProMIS platform to identify epitopes present on misfolded RACK1 and generate antibodies selective for pathogenic, aggregated RACK1. Such antibodies can then be converted into intrabodies with the goal of targeting intracellular, misfolded RACK1 while preserving normal cell function.
20
ProMIS has so far generated 5 mAbs with the desired selectivity (representative example in Fig. 19) and intrabody versions have been generated for testing. Next steps involve generation and screening of additional antibodies, followed by in vitro and in vivo studies for the eventual selection of a candidate(s) for development. Development of intrabodies will be best executed in collaboration with a partner offering gene therapy expertise.
Figure 19
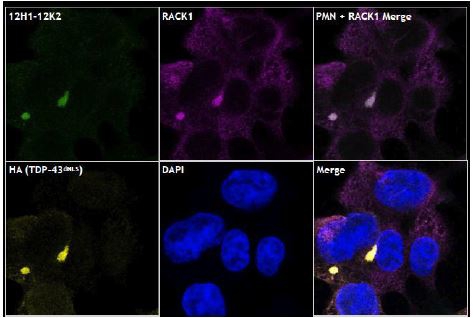
Fig. 19. Selectivity for misfolded RACK1 in aggregates. Representative mAb showing co-localization of staining (green) with RACK1 in TDP-43 aggregates (yellow) but not normal diffuse RACK1 elsewhere in the cytoplasm (purple).
Schizophrenia
DISC1
Protein misfolding and proteostasis have been found to play a role in neurodevelopment disease. DISC1, a candidate for misfolding protein in schizophrenia was first identified in a Scottish family with an autosomal dominant neurodevelopmental syndrome including schizophrenia, which was found to be due to a balanced translocation of part of the C-terminus of a gene subsequently named “disrupted in schizophrenia”. DISC1 is an important hub protein participating in neurogenesis, mitochondrial transport and dynamics in dendrites, cytoskeletal function, and protein translation in adults, especially at the synapse and under conditions of oxidative stress. DISC1 has also been deemed a scaffold protein, and it is possible that proteins of this class are inherently more susceptible to misfolding and aggregation, which is generally associated with both loss-of-function and toxic gain-of-function. However, a true scaffold protein can change its conformation and that of its interactors, and no current evidence exists that the extensive interactome/regulome of DISC1 undergoes conformational change. However, we believe that DISC1 can change conformation in disease, as exemplified by decreased detergent solubility in brains of individuals dying with schizophrenia or psychotic mood disorders and the induced co-aggregation of DISC1 by TDP-43+ inclusions in human frontotemporal dementia. Missense and frameshift mutations in DISC1 have been identified that are linked to familial schizophrenia (R37W, L607F, 4bd deletion extreme 3' end of exon 12), but the role of these mutations has been disputed, and there is no GWAS signal for DISC1 that is statistically significant. However, many variants in interactors of DISC1 show significant association with schizophrenia and cognitive decline. DISC1 itself can oligomerize, which may be associated with toxic gain-of-function as well as loss-of function.
21
Application of the ProMIS platform to DISC1 and its interactome offers the potential to generate selective antibodies to probe the pathobiology of DISC1, gain insight into the biology of schizophrenia and potentially intervene therapeutically. Epitopes predicted to be presented on misfolded DISC1 have been identified and will be used to generate mAbs for further studies.
Future directions
Treatment of neurodegenerative diseases driven by misfolded protein aggregates that exist both extracellularly and intracellularly could potentially benefit from therapeutic modalities that complement our antibody/intrabody approach. We are therefore exploring antisense oligonucleotide (ASO) therapeutic approaches with Dr. Michelle Hastings, Professor and Director of the Center for Genetic Diseases at the Rosalind Franklin University of Medicine and Science, and we are exploring a protein degradation strategy (PROTACS) for ALS with Dr. Justin Yerbury, Professorial Fellow at the School of Chemistry and Molecular Bioscience at the University of Wollongong in Australia.
Alzheimer’s Vaccine Program
The recent development of blood-based biomarkers for neurodegeneration allows for increased screening to potentially diagnose and identify individuals at risk of developing AD. A vaccine capable of inducing an effective antibody response against AβO could therefore be administered prophylactically to at-risk individuals to potentially prevent development of symptomatic disease; and the vaccine could be given therapeutically to individuals living with a diagnosis of AD to potentially inhibit disease progression. ProMIS initiated a program to construct and test a multivalent peptide vaccine candidate for AD containing conformational epitopes identified by the ProMIS platform. The demonstrated ability of these conformational epitopes to induce mAbs with selectivity and protective activity against AβO (see above) supports their potential as vaccine candidates. In addition, the Company believes that a multivalent vaccine containing several B cell epitopes has the potential to maximize “coverage” by immunizing simultaneously against multiple epitopes that can be expressed at variable levels on AβO.
We believe that the same peptide antigens that generate a mAb infusion therapy can be used to create a therapeutic vaccine. The goal of a therapeutic vaccine is to spur the human immune system to generate antibodies that neutralize toxic oligomers, just as the infusion antibodies will hopefully do. The advantage is that a single course of therapy, usually an initial vaccination followed by a booster, can potentially provide years of therapeutic benefit, eliminating the need for frequent costly infusions. The disadvantage of a vaccine approach is that there is no opportunity to improve or refine the antibodies created by the patient’s immune system. With infusion antibodies, the final drug candidate may have gone through a significant amount of refinement and optimization. With therapeutic vaccines, the treatment is only as good as the peptide antigens on which it is based.
In studies conducted by third parties, a first generation vaccine consisting of aggregated human Aβ protein with QS1 adjuvant induced antibody production in AD patients but elicited meningoencephalitis (brain inflammation) and had to be discontinued for safety reasons. Subsequent studies indicated that T helper (“Th”) cell epitopes in the Aβ vaccine gave rise to a pro-inflammatory Th1-type response against the same Aβ epitopes in the brain). The Company believes it can avoid this issue with a vaccine candidate consisting of its AβO B cell epitopes (no Aβ Th epitopes) conjugated to keyhole limpet hemocyanin (KLH) as a carrier protein. KLH has been used in humans and provides Th cell epitopes that are needed to help the development of an antibody response by B cells. Since KLH is a foreign protein not present in human brain, immunization is expected to result in an antibody response against AβO without a potentially detrimental Th cell inflammatory response (Fig. 20). This premise is supported by initial preclinical studies conducted in collaboration with the University of Saskatchewan’s Vaccine and Infectious Disease Organization-International Vaccine Centre (VIDO-InterVac), a global leader in vaccine research and development.
In these studies, 5-6 week old Balb/c mice (n=6/group) received 2 intramuscular (IM) injections (days 0 and 28) of a vaccine candidate construct containing ProMIS’ AβO 301 peptide epitope linked to KLH and formulated with different adjuvants. Analysis of serum samples collected on day 0 and after 1 or 2 vaccinations on days 28 and 48 showed induction of a robust antibody response against the AβO epitope as measured by ELISA (Fig. 21). ELISPOT analysis of spleen cells (immune cells) collected from immunized mice as the end of the study on day 48 showed a lack of Th cell cytokine production in response to stimulation with the AβO epitope thereby indicating that the peptide only contains a B cell epitope. As expected, T cell help was provided by the carrier protein and stimulation with KLH gave rise to the production of Th cytokines. These results support the premise that a vaccine consisting of AβO-restricted conformational B cell epitopes conjugated to KLH for T cell help may successfully induce a protective antibody response against AβO without eliciting a potentially inflammatory Ab- directed Th response. Characterization of immune sera from the mice showed selective binding to AβO compared to Aβ monomers as determined by SPR, and no detectable binding to plaque in brain sections from AD patients as determined by IHC. Optimization of adjuvant formulation and dosing regimen and evaluation of a multivalent construct are ongoing, to be followed by in vivo studies of protective activity in AD mouse models.
22
Figure 20
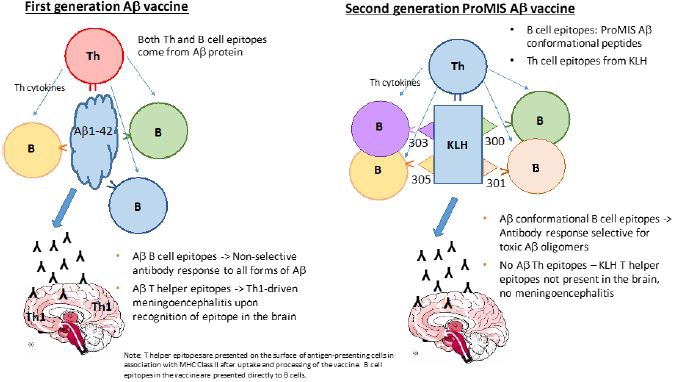
Fig. 20. Illustration of vaccine concept
Figure 21
Fig. 21. Induction of robust antibody response against AβO epitope. Titers of IgG antibodies against the 301 peptide epitope were measured by ELISA. Values for individual mice at baseline and on days 28 and 48 post-immunization are shown.
23
Using the ProMIS discovery platform, our aim is to devise a safe and effective vaccine to induce a specific immune response against toxic AβOs. We have identified a set of six different peptide epitopes selectively exposed on toxic AβOs. Immunization of mice with each of these individual epitopes produced mAbs that selectively bind AβOs, not monomers or fibrils. The immediate goals for this program are to create and evaluate a multivalent AD vaccine comprised of several ProMIS peptide antigens associated with toxic misfolded Aβ.
ProMIS’ Technology Platform and Intellectual Property Portfolio
The basis of ProMIS’ proprietary technology platform is the ability to identify small regions of toxic proteins, including their specific shape or “conformation” that are displayed only on the toxic forms of that protein. We have developed patented methods and know-how combining biology and physics, to identify these small regions of proteins which can be the targets for antibodies. When displayed on the toxic protein, these small regions are known as “epitopes”. ProMIS makes copies of these epitopes, in a precisely defined shape. These drug development tools are called peptide antigens and we believe they are the key to our ability to create antibody therapies, vaccines, and diagnostics.
The ProMIS computational platform consists of two proprietary patented algorithms, the ProMISTM and Collective Coordinates algorithms. Both are molecular dynamics algorithms which combine physics and biology to simulate the folding, or misfolding of proteins. The ProMISTM algorithm was our first to be developed. Collective Coordinates was developed after the ProMISTM algorithm and is the more powerful algorithm for prediction/determination of conformational epitopes. ProMIS has successfully applied these computational algorithms to several misfolded protein categories, looking for epitopes exposed only on a misfolded toxic form which can be used as an antigen to generate an antibody.
Peptide antigens are the key to creating selective antibodies that target toxic misfolded proteins, like our lead therapeutic antibody candidate (PMN310 for AD). PMN310 was created using a peptide antigen that we correctly predicted to be exposed only on toxic AβOs, not the monomeric or plaque forms of Aβ. ProMIS has generated a portfolio of over 20 peptide antigens that have led to selective antibodies against toxic misfolded forms of Aβ for AD, a-syn for MSA and PD, tau for AD, FTLD, PSP, and CBD, TDP-43 and SOD1 for ALS, RACK1 for ALS and HD, and DISC1 for schizophrenia. Those peptide antigens, and the corresponding selective antibodies, represent proprietary reagents that potentially can be used to create proprietary diagnostic tests in neurodegenerative diseases.
Finally, peptide antigens are also a potential key to making vaccines. Therapeutic vaccines are designed to treat a disease by causing the patient’s immune system to make antibodies (or T-Cells, in some areas like cancer) that neutralize the toxic disease driver. The potential advantage of a therapeutic vaccine, if effective, is that a single course of therapy might provide benefit for many years, not requiring frequent, expensive and inconvenient infusions. In preventive therapy, we believe such an approach may be particularly valuable.
24
Overview of ProMIS’ Intellectual Property (IP) Portfolio
The ProMIS IP program consists of a three layered strategy. The first layer of protection comprises two computational algorithms, ProMISTM and Collective Coordinates, obtained under worldwide exclusive license from the UBC. These algorithms are used to predict the specific site and shape (conformation) of epitopes on misfolded proteins implicated in the development of neurodegenerative diseases and on other complex proteins. PCT applications for these disease specific epitopes have been submitted and comprise the second layer of IP protection. Finally, the third layer of protection consists of the composition of matter for the antibodies targeting these disease related epitopes, including use(s) thereof. A summary of the ProMIS patent portfolio as of March 28, 2022 is shown in Appendix A hereto.
Prior Joint Venture Agreements with BC Neuroimmunology Lab Inc.
In July 2020, the Company entered into two collaborative agreements with BC Neuroimmunology Lab Inc. (“BCNI”): the Neurodegen Collaboration and the COVID-19 Collaboration (collectively, the “BCNI Collaborations”). The Company and BCNI entered into the Neurodegen Collaboration to develop and offer highly accurate and objective tests for detection, diagnosis and monitoring of AD. The Company and BCNI entered into the COVID-19 Collaboration to provide a service of highly sensitive and specific serological assays for the detection and characterization of antibodies to the SARS-CoV-2 virus that is responsible for COVID-19. Each of the BCNI Collaborations generally provided for an even split among the Company and BCNI with respect to, among other things, funding, capital expenditures, working capital needs or operating losses, ownership, investment decisions, and cash surpluses. The BCNI Collaborations were both terminated in December 2021 and are no longer material to the Company’s business.
25
The foregoing description of the BCNI Collaborations are qualified in their entirety by reference to the COVID-19 Collaboration and the Neurodegen Collaboration joint venture agreements, which are attached hereto as Exhibits 10.1 and 10.2, respectively.
License Agreements
License Agreement with the University of British Columbia
On February 4, 2009, ProMIS (under its previous name, Amofix Life Sciences Ltd.) entered into an exclusive license agreement with UBC in which ProMIS gained exclusive worldwide rights to develop and commercialize intellectual property rights belonging to UBC, based on its technology relating to misfolded proteins. Such agreement was amended and restated effective October 6, 2015 (as amended and restated, the “UBC License Agreement”). The UBC License Agreement expires on a product by product and country by country basis upon the expiration of ProMIS’ obligation to pay royalties to UBC under the terms thereof (unless terminated earlier pursuant to the terms of the UBC License Agreement). The UBC License Agreement may be terminated by UBC, at its option, upon the occurrence of certain events including, but not limited to, our insolvency, winding up, liquidation or other termination of existence. ProMIS also has the right, in its sole discretion, to terminate the UBC License Agreement upon written notice to UBC. The UBC License Agreement calls for certain customary payments such as an annual license fee and payment to UBC of a low to high single digit royalty on revenues. As of December 31, 2021, the Company has paid a total of $C$175,000 to UBC pursuant to the terms of the UBC License Agreement.
The foregoing description of the UBC License Agreement is qualified in its entirety by reference to the UBC License Agreement, which is attached hereto as Exhibit 10.4.
License Agreement with the University Health Network
On April 4, 2006, ProMIS (under its previous name, Amorfix Life Sciences Ltd.) entered into a license agreement with UHN in which ProMIS obtained an exclusive license to UHN’s ownership rights in SOD1 exposed dimer interface antibody, which was coinvented by Neil Cashman while employed at the University of Toronto and certain employees of UHN (the “Original UHN License Agreement). The parties to the Original UHN License Agreement entered into amendments on July 13, 2006 and July 11, 2007 (together with the Original UHN License Agreement, the “Amended UHN License Agreement”). The Amended UHN License Agreement was amended and restated on November 4, 2013 (together with the Amended UHN License Agreement, the “UHN License Agreement”). Except as otherwise provided in the UHN License Agreement or as mutually agreed to by the parties, the UHN License Agreement expires upon expiration of the last patent issued on any of the technology under the UHN License Agreement. The UHN License Agreement may be terminated upon the occurrence of certain events, including upon the Company’s voluntary petition in bankruptcy or insolvency, the Company’s consent to an involuntary petition in bankruptcy or if a receiving order is given against us, or upon the appointment of a receiver by a court of competent jurisdiction. The UHN License Agreement may also be terminated at the discretion of UHN upon a material breach of the UHN License Agreement by the Company, subject to a period to cure such breach. The Company and UHN may also agree to mutually terminate the UHN License Agreement. The UHN License Agreement calls for certain customary payments such as milestone payments, buyout payments and payment to UHN between a half of a percent to a low single digit royalty on revenues. The aggregate amount of all potential milestone and buyout payments under the UHN License Agreement (excluding royalty payments) is $3,325,000 and, as of December 31, 2021, the Company has paid a total of $C$19,815 to UHN pursuant to the terms of the UHN License Agreement.
The foregoing description of the UHN License Agreement is qualified in its entirety by reference to the UHN License Agreement, which is attached hereto as Exhibits 10.7, 10.7.1, 10.7.2 and 10.7.3.
26
Recent Developments
In addition to those developments discussed elsewhere in this Registration Statement, the following is a summary of the significant developments of the Company that have occurred since March 2020.
On March 24, 2020, ProMIS announced that it had received approval from the TSX to amend the exercise price of an aggregate of 44,182,530 outstanding common share purchase warrants (excluding common share purchase warrants held by Insiders) (the “Repriced Warrants”). The exercise price of the Repriced Warrants was amended to $0.13 per share, effective April 8, 2020 until May 22, 2020. At the end of such period, the Repriced Warrants reverted to their original exercise price. All other terms of the Repriced Warrants remain unchanged.
On September 22, 2020, the Company announced the resignation of Anthony Giovinazzo from the Board. It also announced the initiation of a program to construct and test a multivalent peptide vaccine for AD. The critical first steps in vaccine development will be carried out by VIDO-InterVac, a global leader in vaccine research and development based on the campus of the University of Saskatchewan.
Dr. David Wishart, Distinguished University Professor in the Departments of Biological Sciences and Computing Science at the University of Alberta, was appointed to the Scientific Advisory Board on October 29, 2020. Dr. Wishart has studied protein folding and misfolding for more than 30 years using a combination of computational and experimental approaches. The experimental approaches include NMR spectroscopy, circular dichroism, fluorescence spectroscopy, electron microscopy, protein engineering and molecular biology. The computational methods include molecular dynamics, agent-based modeling, bioinformatics and machine learning. Dr. Wishart has been with the University of Alberta since 1995. He also holds adjunct appointments with the Faculty of Pharmaceutical Sciences and the Department of Pathology and Laboratory Medicine.
On January 1, 2021, James Kupiec resigned as Chief Medical Officer of the Company.
On February 1, 2021, Johannes Roth resigned from the Board.
On March 22, 2021, ProMIS completed a US$7M (C$8.75) private placement of convertible unsecured debentures (the “Debentures”). The Debentures are convertible into Common Shares at the option of the holder at a conversion price of US$0.10 per share and accrue interest at 1% per annum, which is payable annually. At the Company’s election, accrued interest may be paid in cash or Common Shares (such number of shares determined by dividing the interest due by the 5-day volume-weighted average trading price, or “VWAP,” of the Common Shares).
27
The Debentures will mature on March 22, 2026. Prior to the maturity date, the Company may force conversion of the Debentures at the conversion price upon raising US$50 million in equity and/or debt cumulatively. On the maturity date, the Company may redeem the outstanding principal amount of the Debentures in either cash or Common Shares (at the then 5-day VWAP less a 10% discount) or a combination thereof at its election. Amounts redeemed in Common Shares on the maturity date are subject to prior TSX acceptance.
The investors were granted a right to participate, on a pro rata basis, in subsequent Company offerings of equity securities for cash consideration pursuant to a public offering or a private placement.
On May 12, 2021, ProMIS appointed Dr. Rudolph Tanzi, as Chair of its Scientific Advisory Board.
On May 13, 2021, ProMIS appointed Neil Warma to the Board.
On May 21, 2021, the Company announced the initiation of producer cell line development for PMN310, which is the first step in the manufacturing of antibody therapeutics. This program will be carried out by Selexis, SA, using Selexis’ proprietary SUREtechnology Platform™. PMN310, is a humanized mAb that binds with high affinity and selectivity to toxic oligomers of Aβ (a recognized root cause of AD).
On May 27, 2021, ProMIS appointed Dr. David Wishart as Chief Physics Officer.
On August 25, 2021, ProMIS completed a public offering of 125,781,250 units at a price of $0.16 per unit for gross proceeds of $20,125,000. Each unit consisted of one common share and one-quarter purchase warrant. Each warrant entitles the holder thereof to purchase one common share at an exercise price of $0.21 per share at any time for five years.
On September 1, 2021, ProMIS appointed Josh Mandel-Brehm to the Board.
On September 22, 2021, ProMIS appointed Maggie Shafmaster to the Board.
On October 22, 2021, ProMIS appointed Gavin Malenfant as Chief Operating Officer.
On October 22, 2021, Dr. Elliot Goldstein resigned from his role as Chief Executive Officer and President of ProMIS and Eugene Williams accepted the role of Chairman and Chief Executive Officer.
On December 1, 2021, we held our special meeting of shareholders where our shareholders voted and approved the share consolidation resolution, which authorizes the Board, at any time before July 1, 2023, to consolidate our Common Shares within a range from a ratio of thirty pre-consolidation common shares to one post consolidation common share up to a ratio of sixty pre-consolidation common shares for one post-consolidated common share.
On January 18, 2022, ProMIS appointed Dr. Carsten Korth to its Scientific Advisory Board.
On January 27, 2022, ProMIS appointed Dr. Cheryl Wellington to its Scientific Advisory Board.
On February 3, 2022, ProMIS appointed Drs. Guy Rouleau and Alain Dagher to its Scientific Advisory Board.
28
Industry Overview
Markets
ProMIS is applying its patented technology platform to build a portfolio of antibody therapies, therapeutic vaccines, and other antibody based therapies in neurodegenerative diseases and other mis-folded protein diseases, which may include AD, MSA, ALS, PD, LBD, FTLD, PSP, CBD, and schizophrenia. These diseases share a common biologic cause – misfolded versions of proteins, that otherwise perform a normal function, kill neurons and produce disease. ProMIS’ technology platform is an example of the advances in drug discovery enabled by computational power, in silico discovery, and artificial intelligence. We believe this platform provides a potential advantage by selectively targeting the toxic misfolded proteins with therapeutics or detecting them with diagnostics.
Marketing Plans and Milestones
Because ProMIS’ technology potentially has many human and animal applications for both diagnostic and therapeutic uses, its development, marketing and commercial launch schedule must be planned in relation to its available resources. ProMIS intends to out-license the marketing and sales of its product applications to major international healthcare firms for commercial exploitation.
Government Regulations
Regulatory Approval and Certification
All commercial applications of ProMIS’ technology will be subject to substantial regulation and certification in the jurisdictions in which ProMIS or its strategic partners intend to sell its therapeutic products. The initial markets for ProMIS’ product candidates are the U.S. and because the Canadian healthcare (diagnostic and therapeutic) market-place is regulated in a similar manner as in the U.S., ProMIS intends to conform its regulatory and certification scheme to the more rigorous standards imposed by the FDA. Some countries throughout the world provide reciprocal approval based upon the receipt by an innovator of an FDA approval.
Human Therapeutic Products
ProMIS’ human therapeutic product applications will also be subject to rigorous preclinical and clinical testing and other approval procedures by the FDA and similar regulatory agencies in other countries. First, preclinical testing of human therapeutics is conducted on animals in the laboratory to evaluate the potential efficacy, safety and toxicity of a pharmaceutical product candidate. The results of these studies, along with a GMP compliant manufacturing dossier are submitted to the FDA as part of an IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, notifies the applicant of safety concerns or questions related to one or more proposed clinical trials and places the trial on a clinical hold. Typically, the clinical evaluation process involves three phases. In Phase 1, clinical trials are conducted with a small number of healthy human subjects, or in some diseases in patients to determine the early safety profile, the pattern of therapeutic drug distribution and metabolism. The total number of subjects included in Phase 1 clinical trials varies but is generally in the range of 20 to 80. In Phase 2, clinical trials are conducted with groups of patients afflicted with a specific disease to determine preliminary evidence of efficacy, the optimal dosages, and more extensive evidence of safety. Phase 2 clinical trials are typically controlled and conducted in a limited population, usually involving no more than several hundred subjects. In Phase 3, large scale, statistically-driven multi-center, well-controlled clinical trials are conducted with patients afflicted with a target disease in order to provide enough data to demonstrate the efficacy and safety required by the FDA. Phase 3 clinical trials usually involve several hundred to several thousand subjects. In most, though not all, cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to support approval of a drug.
Data from clinical trials conducted outside the U.S. may be accepted by the FDA subject to certain conditions. For example, the clinical trial must be conducted in accordance with Good Clinical Practices (“GCP”) requirements and/or the FDA must be able to validate the data from the clinical trial through an onsite inspection if it deems such inspection necessary. Where data from foreign clinical trials are intended to serve as the sole basis for marketing approval in the U.S., the FDA will not approve the application on the basis of foreign data alone unless those data are considered applicable to the U.S. patient population and U.S. medical practice, the clinical trials were performed by clinical investigators of recognized competence, and the data is considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
29
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of a New Drug Application (“NDA”) or Biologics License Application (“BLA”) requesting approval to market the product for one or more indications. In most cases, the submission of an NDA or BLA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA or BLA, for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA or BLA is submitted to FDA because the FDA has approximately two months to make a “filing” decision.
The FDA conducts a preliminary review of all NDAs or BLAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA or BLA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA or BLA to determine, among other things, whether the drug or biologic is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality, and purity.
The FDA may refer an application for a novel drug or biologic to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, which reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to ensure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical trial sites to assure compliance with current good clinical practice, or cGCP, requirements.
After evaluating the NDA or BLA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA or BLA and may require additional clinical or preclinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
30
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s or biologic’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a Risk Evaluation and Mitigation Strategy, or REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Expedited Development and Review Programs
The FDA maintains several programs intended to facilitate and expedite development and review of new drugs or biologics to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These programs include Fast Track designation, Breakthrough Therapy designation, Priority Review and Accelerated Approval, and the purpose of these programs is to either expedite the development or review of important new drugs to get them to patients more quickly than standard FDA review timelines typically permit.
A drug is eligible for Fast Track designation if it is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address unmet medical needs for such disease or condition. Fast Track designation provides increased opportunities for sponsor interactions with the FDA during preclinical and clinical development, in addition to the potential for rolling review once a marketing application is filed. Rolling review means that the agency may review portions of the marketing application before the sponsor submits the complete application. In addition, a drug may be eligible for Breakthrough Therapy designation if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough Therapy designation provides all the features of Fast Track designation in addition to intensive guidance on an efficient drug development program, and FDA organizational commitment to expedited development, including involvement of senior managers and experienced review staff in a cross-disciplinary review, where appropriate.
Any product submitted to the FDA for approval, including a product with Fast Track or Breakthrough Therapy designation, may also be eligible for additional FDA programs intended to expedite the review and approval process, including Priority Review designation and Accelerated Approval. A product is eligible for Priority Review designation, once an NDA or BLA is submitted, if the drug that is the subject of the marketing application has the potential to provide a significant improvement in safety or effectiveness in the treatment, diagnosis or prevention of a serious disease or condition. Under priority review, the FDA’s goal date to take action on the marketing application is six months compared to ten months for a standard review. Products are eligible for Accelerated Approval if they can be shown for a serious or life-threatening condition and to have an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, which is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
Accelerated Approval is usually contingent on a sponsor’s agreement to conduct additional post-approval studies to verify and describe the product’s clinical benefit. The FDA may withdraw approval of a drug or an indication approved under Accelerated Approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product, other evidence demonstrates that the product is not shown to be safe and effective under conditions of use, or required post-approval studies are not conducted with due diligence. In addition, the FDA generally requires, as a condition for Accelerated Approval, that all advertising and promotional materials intended for dissemination or publication within 120 days of marketing approval be submitted to the agency for review during the pre-approval review period. After the 120-day period has passed, all advertising and promotional materials must be submitted at least 30 days prior to the intended time of initial dissemination or publication.
31
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or the time period for FDA review or approval may not be shortened. Furthermore, Fast Track designation, Breakthrough Therapy designation, Priority Review and Accelerated Approval do not change the scientific or medical standards for approval or the quality of evidence necessary to support approval, though they may expedite the development or review process.
Post-Approval Requirements
Drugs or biologics manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There are continuing, annual user fee requirements for any marketed products and the establishments where such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA or BLA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug and biologic manufacturers and other entities involved in the manufacture and distribution of approved drugs or biologics are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval of a drug or biologic is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs or biologics may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In many foreign countries, drugs and biologics are subject to regulatory requirements in addition to and sometimes different than the U.S. requirements described herein.
32
Companion Diagnostics
The FDA defines in vitro, or IVD, companion diagnostic device as an in vitro diagnostic device that provides essential information for the safe and effective use of a corresponding therapeutic product. The use of an IVD companion diagnostic device with a therapeutic product is stipulated in the instructions for use in the labeling of both the diagnostic device and the corresponding therapeutic product, including the label. Applications for an IVD companion diagnostic device and its corresponding therapeutic product will be reviewed and approved according to applicable regulatory requirements. The IVD companion diagnostic device application will be reviewed and approved or cleared under the device authorities of the Federal Food, Drug, and Cosmetic Act (“FD&C Act”) and relevant medical device regulations; the therapeutic product application will be reviewed and approved under section 505 of the FD&C Act (i.e., drug products) or section 351 of the Public Health Service Act (i.e., biological products) and relevant drug and biological product regulations. The FDA intends to review each IVD companion diagnostic device submission within the context of, or in conjunction with, its corresponding therapeutic product, and FDA review of the IVD companion diagnostic device and the therapeutic product will be carried out collaboratively among relevant FDA offices.
Ideally, a therapeutic product and its corresponding IVD companion diagnostic device should be developed contemporaneously, with the clinical performance and clinical significance of the IVD companion diagnostic device established using data from the clinical development program of the corresponding therapeutic product. Some of our current and future product development candidates may depend upon co-development of accurate genetic and potentially other IVDs. Thus, we will likely need to comply with both FDA drug and medical device regulations. This adds additional cost and complexity to our development programs. The availability of IVD companion diagnostics can allow more efficient development programs and more appropriate use of products in the marketplace with more predictable outcomes for patients and higher value medicines. Ultimately FDA approval of the IVD will be required to allow approval of some of our products. However, technical difficulties or other issues could delay or disrupt the development of our products.
U.S. Healthcare Fraud and Abuse Laws and Compliance Requirements
We are subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales and marketing programs for drugs and biologics. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect such operations include:
| · | the federal Anti-Kickback Statute is a criminal statute which prohibits, among other things, persons from soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value; |
| · | federal false claims and civil monetary penalties laws, including the federal civil False Claims Act, which prohibits anyone from, among other things, knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services that are false or fraudulent; |
| · | provisions of federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created federal criminal statutes that prohibit, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program or making false statements in connection with the delivery of or payment for healthcare benefits, items or services. In addition, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”) and its implementing regulations, impose certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| · | the federal Physician Payments Sunshine Act requirements, under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the Affordable Care Act, which require manufacturers of certain drugs and biologics to track and report to CMMS payments and other transfers of value they make to U.S. physicians and teaching hospitals as well as physician ownership and investment interests in the manufacturer; and |
33
| · | the Foreign Corrupt Practices Act of 1977 (“FCPA”) which prohibits U.S. businesses and their representatives from offering to pay, paying, promising to pay or authorizing the payment of money or anything of value to a foreign official in order to influence any act or decision of the foreign official in his or her official capacity or to secure any other improper advantage in order to obtain or retain business. |
Environmental Regulation
The Company may also be subject to foreign and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. There can be no assurance that the Company will not incur significant costs to comply with laws and regulations in the future or that such laws or regulations will not have a material adverse effect upon the Company’s business, financial condition and results of operations.
Pricing and Reimbursement
Precision therapeutic products and their accompanying companion diagnostic are largely paid for based on third party payor reimbursement. In the U.S., concurrent with approval for commercialization of such therapeutic products by the FDA, each therapeutic product is assigned a product code, and its associated companion diagnostic assigned a similar code, or CPT. Each product code and CPT is then assigned a reimbursement level by the CMMS. Third party insurance payors typically establish a specific fee to be paid for each code submitted. Third party payor reimbursement policies are generally determined with reference to the reimbursement for CPT codes for Medicare patients which themselves are determined on a national basis by CMMS.
In recent years, Congress has considered reductions in Medicare reimbursement levels for drugs administered by physicians. CMMS, the agency that administers the Medicare and Medicaid programs, also has authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products. While Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Parallel to this regulatory reimbursement scheme in the U.S., other countries also regulate reimbursement similar to the U.S. Therefore, it is important that ProMIS establish for its human diagnostic and therapeutic products reimbursement schemes, which provide ultimate financial payment for ProMIS’ products consistent with its business plan.
Healthcare Reform Measures
The U.S. and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system. The U.S. government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
34
The Affordable Care Act substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum Medicaid rebate for both branded and generic drugs, expanded the 340B program, and revised the definition of average manufacturer price (“AMP”), which could increase the amount of Medicaid drug rebates manufacturers are required to pay to states. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service Medicaid utilization, to include the utilization of Medicaid managed care organizations as well and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the number of rebates due on those drugs. On February 1, 2016, CMMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016. Since that time, there have been significant ongoing efforts to modify or eliminate the Affordable Care Act.
The Affordable Care Act has been subject to challenges in the courts. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional and remanded the case to the Texas District Court to reconsider its earlier invalidation of the entire Affordable Care Act. An appeal was taken to the U.S. Supreme Court. On June 17, 2021, the U.S. Supreme Court ruled that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the U.S. Supreme Court did not rule on the constitutionality of the Affordable Care Act or any of its provisions.
Other legislative changes have been proposed and adopted since passage of the Affordable Care Act. The Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of an amount greater than $1.2 trillion for the fiscal years 2012 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year. The Bipartisan Budget Act of 2018 retained the federal budget “sequestration” Medicare payment reductions of 2%, and extended it through 2027 unless congressional action is taken, and also increased labeler responsibility for prescription costs in the Medicare Part D coverage gap. On January 2, 2013, the American Taxpayer Relief Act was signed into law, which, among other things, reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Further legislative and regulatory changes under the Affordable Care Act remain possible, although the Biden Administration has signaled that it plans to build on the Affordable Care Act and expand the number of people who are eligible for subsidies under it. President Biden indicated that he intends to use executive orders to undo changes to the Affordable Care Act made by the Trump administration and would advocate for legislation to build on the Affordable Care Act. It is unknown what form any such changes or any law would take, and how or whether it may affect our business in the future. We expect that changes or additions to the Affordable Care Act, the Medicare and Medicaid programs, allowing the federal government to directly negotiate drug prices and changes stemming from other healthcare reform measures, especially with regard to healthcare access, financing or other legislation in individual states, could have a material adverse effect on the healthcare industry.
We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, if approved, or additional pricing pressures.
35
Regulation Outside of the U.S.
In addition to regulations in the U.S., we may be subject to a variety of regulations in foreign jurisdictions that govern, among other things, clinical trials and any commercial sales and distribution of our products, if approved, either directly or through our distribution partners. Whether or not we obtain FDA approval for a product candidate, we must obtain the requisite approvals from regulatory authorities in foreign jurisdictions prior to the commencement of clinical trials or marketing and sale of the product in those countries. The foreign regulatory approval process and the time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Some foreign jurisdictions have a drug product approval process similar to that in the U.S., which requires the submission of a clinical trial application much like the IND prior to the commencement of clinical studies. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others. Moreover, some nations may not accept clinical studies performed for U.S. approval to support approval in their countries or require that additional studies be performed on natives of their countries. In addition, in certain foreign markets, the pricing of drug products is subject to government control and reimbursement may in some cases be unavailable or insufficient. We face the risk that the resulting prices would be insufficient to generate an acceptable return to us or any future partner of ours. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, and criminal prosecution.
Commercial Marketing Plans and Strategies
ProMIS does not intend to market its therapeutic products and companion diagnostics it develops that require extensive distribution channels. Instead, ProMIS intends to license to, or enter into strategic alliances with, larger pharmaceutical entities that are equipped to manufacture and/or market ProMIS’ products through their distribution networks. ProMIS may license some or all of its patent rights to more than one company to achieve the fullest development, marketing and distribution of its products. To this end ProMIS intends to continue to develop and improve its proprietary technologies and to expand the applications of its technologies in the healthcare markets.
Generate Product Revenues
Revenues, if any, from its precision therapeutics pipeline and companion diagnostics are expected to be generated from research funding, license fees, milestone payments, co-development funding, and royalties from partnerships to be completed by ProMIS with selected third-party, multi-national health care firms. As of the date of this form, ProMIS has not generated any significant product revenues.
Develop Collaborative Customer-Funded Commercialization Agreements
In order to increase market exposure of its products and to capitalize on a partner’s clinical development competencies, market position, and distribution capabilities, ProMIS intends to develop its projects with collaborative commercial partners who will fund further product development projects incorporating ProMIS’ technology. These collaborative arrangements typically will provide for a jointly funded development project and contemplate a licensing arrangement (which may be entered into at the same time as the development project or at a later date) under which, if a project is commercialized by the collaborative partner, ProMIS would potentially receive license fees, royalty payments from product sales and manufacturing revenue. ProMIS believes that such arrangements with major commercial partners will serve to validate its proprietary technologies in human healthcare areas and thereby assist ProMIS in attracting additional licensing arrangements on favorable terms.
36
Enhance Out-licensing of ProMIS Requirements
Where practical, ProMIS will outsource its product manufacturing and has explored and will continue to evaluate the possibility of entering into strategic manufacturing alliances with appropriate third parties.
Competition
Human Healthcare Products Competition
ProMIS will compete with many large and small pharmaceutical companies that are developing and/or marketing therapeutic compounds for AD, ALS and/or PD. Many large pharmaceutical companies and smaller biotechnology companies maintain well-funded research departments concentrating on therapeutic approaches to neurodegenerative diseases. ProMIS expects substantial competition from these companies as they develop different and/or novel approaches to the treatment of these diseases. Some of these approaches may directly compete with the technology that ProMIS is currently developing.
If approved, PMN310 will compete with therapies currently approved for the treatment of patients with AD, which have primarily been developed to treat the symptoms of AD rather than the underlying cause of the disease, such as memantine and cholinesterase inhibitors. PMN310 may also compete with one or more potentially disease-modifying therapeutics that target Aβ or amyloid plaques. Biogen’s aducanumab was approved by the FDA in June 2021 under the accelerated approval pathway, which allows for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint.
Eisai has disclosed that the pivotal trial program for Lecanemab will conclude in September of 2022. If successful, it may be the next approved anti-amyloid therapy. In many therapeutic categories, after initial approvals validate a general mechanistic approach, competitive dynamics are driven by relative safety, efficacy, convenience, and cost effectiveness. We expect this will be the case in the anti-amyloid immunotherapy category
Other companies known to be developing therapies with Aβ/amyloid plaque-related targets include Alzheon, Inc., Alzinova AB, Chugai Pharmaceutical Co. Ltd., Cognition Therapeutics, Inc., Eisai Co., Ltd., Eli Lilly and Company, Grifols, S.A., KalGene Pharmaceuticals, Inc., Neurimmune AG, Novartis AG, Acumen Pharmaceuticals Inc., Prothena Biosciences, Inc., Roche Holding AG (including Genentech, its wholly owned subsidiary) and Wren Therapeutics, Inc. Additionally, PMN310, if approved, may also compete with other potential therapies intended to address underlying causes of AD that are being developed by several companies, including AbbVie Inc., AC Immune SA, Alector, Inc., Anavex Life Sciences Corp., Annovis Bio, Inc., Athira Pharma, Inc., Biohaven Pharmaceuticals, Inc., Cassava Sciences, Inc., Cortexyme, Inc., Denali Therapeutics, Inc., Johnson & Johnson (including Janssen, its wholly-owned subsidiary) and Takeda Pharmaceutical Co. Ltd. Some of these competitors are developing therapies that either seek to block the aggregation of amyloid oligomers (for example, Alzheon, Inc.), or mitigate the toxicity of amyloid oligomers (for example, Cognition Therapeutics, Inc.). These and other therapies may end up being used as complementary therapies in clinical practice, in addition to antibodies targeting aggregated amyloid.
In the intense competitive environment that is the human pharmaceutical industry, those companies that complete clinical trials, obtain regulatory approval and commercialize their therapeutic products first may enjoy competitive advantages. ProMIS believes that it will develop compounds with characteristics that may enable them, if fully developed, to have a market impact. A number of major human pharmaceutical companies have significant programs to develop drugs for the treatment of neurodegenerative disease. These companies include Eisai/Pfizer, Novartis, Merck, Genentech, Lilly, Biogen, Amgen and Johnson & Johnson.
Proprietary Protection
ProMIS has acquired the rights to certain proprietary discovery platforms for the identification of proteins involved in misfolding diseases embodied in various national and international patent applications. ProMIS has also filed international patent applications related to immunotherapy targeting toxic forms of SOD1 and TDP-43 for ALS, toxic oligomers of Aβ for AD and toxic aggregates for a-syn for PD to further protect its intellectual property rights related to its therapeutic programs. In addition, the Company has obtained proprietary rights to two computational algorithms (ProMISTM and Collective Coordinates) for identification of DSEs in protein misfolding diseases as well as predicted DSEs against multiple disease targets. ProMIS intends to aggressively protect the commercial applications for diagnostic, therapeutic and prophylactic applications of these discoveries. In addition, ProMIS has developed know-how, which it may elect to keep as trade secrets and not publicly disclose in patent applications.
Employees
ProMIS seeks to hire qualified scientists and key employees as needed. As of March 25, 2022, the Company had six total employees, consisting of five full-time employees and one part-time employee. The remainder of the scientists and key personnel had consulting agreements with ProMIS.
The following specific factors could materially adversely affect the Company and should be considered when deciding whether to make an investment in the Company. The risks and uncertainties described in this Registration Statement and the information incorporated by reference herein are those we currently believe to be material, but they are not the only ones we face. Additionally, investors should not interpret the disclosure of a risk to imply that the risk has not already materialized. If any of the following risks, or any other risks and uncertainties that we have not identified or that we currently consider not to be material, actually occur or become material risks, our business, prospects, financial condition, results of operations and cash flows, and consequently the price of the Common Shares could be materially and adversely affected. In all these cases, the trading price of our securities could decline, and prospective investors could lose all or part of their investment.
Investors should carefully consider the risk factors set out below and consider all other information contained herein and in the Company’s other public filings before making an investment decision.
Risks Related to the COVID-19 Pandemic
Our business and operations have and may be further adversely affected by the evolving and ongoing COVID-19 global pandemic.
Our business and operations have and may be further adversely affected by the effects of the recent and evolving COVID-19 virus, and the efforts to mitigate it, which was declared a global pandemic by the World Health Organization in March 2020. The COVID-19 pandemic has resulted in travel and other restrictions in order to reduce the spread of the disease, including public health directives and orders in the U.S. and the European Union that, among other things and for various periods of time, directed individuals to shelter at their places of residence, directed businesses and governmental agencies to cease non-essential operations at physical locations, prohibited certain non-essential gatherings and events and ordered cessation of non-essential travel. For example, in April 2020, the COVID-19 pandemic caused us to close one of our labs for several months in order to comply with local and federal mandates aimed at preventing the further spread of the virus. Future remote work policies and similar government orders or other restrictions on the conduct of business operations related to the COVID-19 pandemic may negatively impact productivity and may disrupt our ongoing research and development activities and our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. Further, such orders also may impact the availability or cost of materials, which would disrupt our supply chain and manufacturing efforts and could affect our ability to conduct ongoing and planned clinical trials and preparatory activities. We have also experienced preclinical supply chain disruptions, including increased prices and reduced availability of manufacturing materials and supplies related to our IND-enabling work as well as for non-human primates for our nonclinical studies, as a result of the COVID-19 pandemic. Additionally, business interruptions to external parties, such as academic institutions or potential pharmaceutical collaborators, caused by the COVID-19 pandemic may impact our ability to progress or effectively partner our programs.
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, the continued widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our Common Shares.
37
The global COVID-19 pandemic continues to rapidly evolve. The extent to which the COVID-19 pandemic impacts our business and operations, including our clinical development and regulatory efforts, will depend on future developments that are highly uncertain and cannot be predicted with confidence as of the filing of this Registration Statement, such as the ultimate geographic spread of the disease, the duration of the outbreak, the duration and effect of business disruptions and the short-term effects and ultimate effectiveness of travel restrictions, quarantines, social distancing requirements and business closures in the U.S. and other countries to contain and treat the disease. Accordingly, we do not yet know the full extent of potential delays or impacts on our business, our clinical and regulatory activities, healthcare systems or the global economy as a whole. However, these impacts could adversely affect our business, financial condition, results of operations and growth prospects.
In addition, to the extent the ongoing COVID-19 pandemic adversely affects our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described in this “Risk Factors” section.
The ongoing COVID-19 pandemic may negatively impact the availability of scientific staff, physicians and other healthcare professionals, which may negatively impact our business, financial condition and results of operations.
We rely on the availability of scientific staff, physicians and other healthcare professionals to provide testing services. If scientific staff, physicians and other healthcare professionals were unable or unwilling to provide these services in the future due to any reason including infection due to COVID-19, this would cause interruptions in our business until mitigated accordingly. As such, vacancies and disabilities relating to our current staff may cause interruptions in our business and result in lower revenues.
As we expand our operations, we may encounter difficulty in securing the necessary professional scientific, medical and skilled support staff to support our expanding operations, which may adversely affect our business, financial condition and results of operations. Additionally, we follow posted health guidelines, as and when posted, to protect the health of our employees and decrease the potential impact of serious illness, including COVID- 19, on our operations. However, should an employee of, or visitor to, our offices or research and development facilities become infected with COVID-19, it could place our workforce at risk, which could result in the disruption or suspension of operations at our facilities. Such a suspension in operations could also be mandated by governmental authorities in response to the COVID-19 pandemic. This would negatively impact our operations which could adversely impact our business, financial condition and results of operations.
Risks Related to the Development of Our Product Candidates
Our product candidates are still in the early stages of development and there is significant uncertainty that any such products will actually be developed.
Our product candidates are at an early stage of development. Significant additional investment in research and development, product validation, technology transfer to manufacturing, production scale-up, manufacturing, clinical testing, and regulatory submissions of such product candidates is required prior to commercialization. There can be no assurance that any such product candidates will actually be developed and, if developed, will be approved. The development and regulatory processes may require access to rare biofluid and tissue samples from people and animals which may not be available to us in sufficient amounts or in a timely fashion to allow us to complete the development or receive regulatory approval of any product candidate or process. A commitment of substantial time and resources is required to conduct research and clinical trials if we are to complete the development of any product candidate. It is not known whether any of these product or process candidates will meet applicable health regulatory standards and obtain required regulatory approvals, or whether such products, if approved, can be produced in commercial quantities at reasonable costs and be successfully marketed, or if our investment in any such products will be recovered through sales or royalties.
We expect to incur substantial capital expenditures in connection with the development of our product candidates. If we fail to successfully develop and sell all or any of our product candidates, then we will not earn any return on our investment in these future products, which will adversely affect our results of operations and could adversely affect the market price of the Common Shares. Our success in developing and selling new products will depend upon multiple factors, including:
| · | our ability to develop safe and effective products; |
| · | our serology assays and vaccines achieving the desired sensitivity for antibody-based immunity and immune response, as applicable; |
| · | acceptance of the product by the medical community and by patients and third-party payors; |
| · | inherent development risks, such as the product proving to be unsafe or unreliable, or not having the anticipated effectiveness; and |
| · | our ability to develop repeatable processes to manufacture new products in sufficient quantities. |
If any of these factors cannot be overcome, we may not be able to develop and introduce our products in a timely or cost-effective manner, which could adversely affect our future growth and results of operations. Our failure to develop and obtain approval of our product candidates could adversely affect the market price of the Common Shares.
38
We have concentrated a portion of our research and development efforts on the treatment of AD, a field that has seen very limited success in drug development.
We have focused our research and development efforts on developing effective treatments for AD. Collectively, efforts by pharmaceutical companies in the field of AD have seen very limited successes in drug development. There are few approved products available for patients with AD. Only one disease-modifying therapeutic option has been approved by the FDA. Biogen’s Aduhelm (aducanumab), a mAb administered via infusion, received accelerated approval from the FDA on June 7, 2021. We cannot be certain that our approach will lead to the development of approvable or marketable products. With the exception of Aduhelm, the only drugs approved by the FDA to treat patients with AD address the symptoms of the disease. Since 2003, over 500 clinical studies have been completed and only Aduhelm has been approved by the FDA, compared to a success rate of 50% to 80% for all other drug candidates. As a result, the FDA has a limited set of products to rely on in evaluating PMN310. This could result in a longer than expected regulatory review process, increased expected development costs or the delay or prevention of commercialization of PMN310 for the treatment of AD.
Our business is heavily dependent on the successful development, regulatory approval and commercialization of PMN310 and any future product candidates that we may develop or acquire, including PMN442 and PMN267.
We currently have no products approved for sale, and our lead product candidate is in early stages of development. The success of our business, including our ability to finance our company and generate revenue in the future, will primarily depend on the successful development, regulatory approval and commercialization of our product candidates and, in particular, the advancement of PMN310. However, given our stage of development, it may be many years, if we succeed at all, before we have demonstrated the safety and efficacy of a product candidate sufficient to warrant approval for commercialization. We cannot be certain that our product candidates will receive regulatory approval or be successfully commercialized even if we receive regulatory approval.
The clinical and commercial success of PMN310 and any future product candidates that we may develop or acquire will depend on a number of factors, including the following:
| · | our ability to raise any additional required capital on acceptable terms, or at all; |
| · | our ability to complete an IND enabling studies and successfully submit INDs or comparable applications; |
| · | timely completion of our preclinical studies and clinical trials, which may be significantly slower or cost more than we currently anticipate and will depend substantially upon the performance of third-party contractors; |
| · | delays or difficulties in enrolling and retaining patients in our clinical trials; |
| · | whether we are required by the FDA or similar foreign regulatory agencies to conduct additional clinical trials or other studies beyond those planned to support the approval and commercialization of our product candidates or any future product candidates; |
| · | acceptance of our proposed indications and primary endpoint assessments relating to the proposed indications of our product candidates by the FDA and similar foreign regulatory authorities; |
| · | our ability to demonstrate to the satisfaction of the FDA and similar foreign regulatory authorities the safety, efficacy and acceptable risk to benefit profile of our product candidates or any future product candidates; |
39
| · | the prevalence, duration and severity of potential side effects or other safety issues experienced with our product candidates or future approved products, if any; |
| · | achieving and maintaining, and, where applicable, ensuring that our third-party contractors achieve and maintain, compliance with our contractual obligations and with all regulatory requirements applicable to our product candidates or any future product candidates or approved products, if any; |
| · | the ability of third parties with whom we contract to manufacture adequate clinical trial and commercial supplies of our product candidates or any future product candidates remain in good standing with regulatory agencies and develop, validate and maintain commercially viable manufacturing processes that are compliant with cGMPs; |
| · | the convenience of our treatment or dosing regimen; |
| · | the timely receipt of necessary marketing approvals from the FDA and similar foreign regulatory authorities; |
| · | acceptance by physicians, payors and patients of the benefits, safety and efficacy of our product candidates or any future product candidates, if approved, including relative to alternative and competing treatments; |
| · | the willingness of physicians, operators of clinics and patients to utilize or adopt any of our product candidates or any future product candidates, if approved; |
| · | our ability to achieve sufficient market acceptance, coverage and adequate reimbursement from third-party payors and adequate market share and revenue for any approved products; |
| · | the COVID-19 pandemic, which may result in clinical site closures, delays to patient enrollment, patients discontinuing their treatment or follow up visits or changes to trial protocols; |
| · | our ability to successfully develop a commercial strategy and thereafter commercialize our product candidates or any future product candidates in the U.S. and internationally, if approved for |
| · | marketing, reimbursement, sale and distribution in such countries and territories, whether alone or in collaboration with others; |
| · | patient demand for our product candidates, if approved, including patients’ willingness to pay out-of-pocket for any approved products in the absence of coverage and/or adequate reimbursement from third-party payors; |
| · | our ability to establish and enforce intellectual property rights in and to our product candidates or any future product candidates; and |
| · | our ability to avoid third-party patent interference, intellectual property challenges or intellectual property infringement claims. |
In addition, the FDA or other regulatory agencies may not agree with our clinical development plan and require that we conduct additional clinical trials to support our regulatory submissions. We have not yet conducted an end of Phase 2 meeting with the FDA to discuss the registration pathway for PMN310, and our current clinical development plans for PMN310 in AD may change as a result of future interactions with the FDA. For example, the FDA may not accept the results of the ongoing PMN310 clinical trials and may require that we conduct additional trials, including more than one pivotal trial, in order to gain approval in AD. Furthermore, any approval of PMN310 for AD may be limited to PMN310 in combination with the existing standard of care.
40
These factors, many of which are beyond our control, could cause us to experience significant delays or an inability to obtain regulatory approvals or commercialize our product candidates. Even if regulatory approvals are obtained, we may never be able to successfully commercialize any of our product candidates. Accordingly, we cannot provide assurances that we will be able to generate sufficient revenue through the sale of our product candidates or any future product candidates to continue our business or achieve profitability.
Our approach to the potential treatment of AD is based on a novel therapeutic approach, which exposes us to unforeseen risks.
There is no current scientific or general consensus on the causation of AD or method of action to treat AD. We have discovered and are developing PMN310, a humanized antibody that selectively targets AβO, or AβOs, to treat AD. Our approach is based on research on AβOs, globular assemblies of the Aβ peptide that are distinct from other forms of amyloid. AβOs have gained scientific acceptance as primary toxins involved in the initiation and propagation of AD pathology. Based on the results of our nonclinical studies to date, we believe PMN310 is different from current and prior clinical-stage anti-amyloid drugs and product candidates based on its selectivity for AβOs. We believe that this is a novel mechanism which has the potential to provide more favorable outcomes, as compared to approved therapies and product candidates in development and may potentially slow disease progression. However, we may ultimately discover that PMN310 does not possess properties required for therapeutic effectiveness. We have no evidence regarding the efficacy, safety or tolerability of PMN310 in humans. We may spend substantial funds attempting to develop PMN310 or other product candidates and never succeed in doing so.
The market for any products that we successfully develop, if any, will also depend on the cost of the product. We do not yet have sufficient information to reliably estimate what it would cost to commercially manufacture PMN310, if approved, and the actual cost to manufacture PMN310 or any drug we develop in the future could materially and adversely affect the commercial viability of the drug. We may also find that the manufacture of our product candidates is more difficult than anticipated, resulting in an inability to produce a sufficient amount of our product candidates for our clinical trials or, if approved, commercial supply. If we do not successfully develop PMN310 or any other drug we develop with drug product cannot be reliably and economically manufactured at scale, we will not become profitable, which would materially and adversely affect the value of our Common Shares.
We may not successfully expand our pipeline of product candidates, including by pursuing additional indications for PMN310 or by in-licensing or acquiring additional product candidates for other diseases.
A key element of our strategy is to build and expand our pipeline of product candidates, including by developing PMN310 for the treatment AD, and by identifying other product candidates. In addition, we may in-license or acquire additional product candidates for other diseases. We may not be able to identify or develop additional product candidates that are safe, tolerable and effective. Even if we are successful in continuing to build our pipeline, the potential product candidates that we identify, in-license or acquire may not be suitable for clinical development. For example, our research methodology may be unsuccessful in identifying potential drug candidates or those we identify may be shown to have harmful side effects or other characteristics that make them unmarketable or unlikely to receive regulatory approval. We cannot guarantee that we will be successful in identifying additional potential drug candidates, or that we will be able to successfully identify and in-license new and valuable product candidates from other parties.
41
Nonclinical and clinical drug development involves a lengthy, expensive and uncertain process. The results of nonclinical studies and early clinical trials are not always predictive of future results. PMN310 or any other product candidate that we advance into clinical trials may not achieve favorable results in later clinical trials, if any, or receive marketing approval.
The research and development of product candidates is extremely risky. Only a small percentage of product candidates that enter the development process ever receive marketing approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete nonclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain.
The results of nonclinical studies and early clinical trials are not necessarily predictive of future results and PMN310, or any other product candidate that we may develop, may not be further developed or have favorable results in later studies or trials. Clinical trial failure may result from a multitude of factors including, but not limited to, flaws in study design, dose selection, placebo effect, patient enrollment criteria and failure to demonstrate favorable safety or efficacy traits. As such, failure in clinical trials can occur at any stage of testing. A number of companies in the pharmaceutical industry have suffered setbacks in the advancement of their product candidates into later-stage clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding results in earlier nonclinical studies or clinical trials. In addition, the results of clinical trials in one set of patients or disease indications may not be predictive of those obtained in another. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among clinical trial participants. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. This is particularly true in AD, where failure rates historically are higher than in most other disease areas.
In the event of negative or inconclusive results, we may decide, or regulatory authorities may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from clinical trials and nonclinical studies is susceptible to varying interpretations, and regulatory authorities may not interpret our data as favorably as we do, which may further delay, limit or prevent development efforts, clinical trials or marketing approval. Furthermore, as more competing product candidates within a particular class of drugs proceed through clinical development to regulatory review and approval, the amount and type of clinical data that may be required by regulatory authorities may increase or change.
If we are unable to complete nonclinical studies or clinical trials of PMN310 or future product candidates, due to safety concerns or otherwise, or if the results of these trials are not sufficient to convince regulatory authorities of their safety or efficacy, we will not be able to obtain marketing approval for commercialization on a timely basis or at all. Even if we are able to obtain marketing approval for PMN310 or any future product candidates, those approvals may be for indications or dose levels that deviate from our desired approach or may contain other limitations that would adversely affect our ability to generate revenue from sales of those product candidates. Moreover, if we are not able to differentiate our product candidate against other approved product candidates within the same class of drugs, or if any of the other circumstances described above occur, our business would be harmed and our ability to generate revenue from that class of drugs would be severely impaired.
Clinical failure can occur at any stage of clinical development and we have never completed a clinical trial or submitted NDA, BLA, or marketing authorization application, or MAA.
We are early in our development efforts for PMN310, and will need to successfully complete our ongoing and planned clinical trials, including pivotal clinical trials, in order to obtain FDA approval to market PMN310 or any other product candidate we seek to develop. Carrying out clinical trials and the submission of a successful BLA is a complicated process. Although members of our team have significant experience in clinical development of drugs through regulatory approval, as an organization, we have just begun conducting our first clinical trial, we have no experience in conducting any clinical trials, we have limited experience in preparing regulatory submissions and we have not previously submitted a BLA for any product candidate.
42
In addition, we have had limited interactions with the FDA and cannot be certain how many clinical trials of PMN310 will be required or how such trials should be designed. Consequently, we may be unable to successfully and efficiently execute and complete necessary clinical trials in a way that leads to BLA submission and approval of PMN310 or any other product candidate. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop. Failure to commence or complete, or delays in, our planned clinical trials, could prevent us from or delay us in commercializing PMN310 or any future product candidates we may develop, and failure to successfully complete any of these activities in a timely manner could have a material adverse impact on our business and financial performance.
We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval or commercialize our product candidates, including:
| · | regulatory authorities, Institutional Review Boards, or IRBs, or Ethics Committees, or ECs, may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site or we may fail to reach a consensus with regulatory authorities on trial design; |
| · | regulatory authorities in jurisdictions in which we seek to conduct clinical trials may differ from each other on our trial design, and it may be difficult or impossible to satisfy all such authorities with one approach; |
| · | we may have delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different contract research organizations, or CROs, and trial sites; |
| · | we may be unable to add or be delayed in adding a sufficient number of clinical trial sites and obtaining IRB or independent EC approval at each clinical trial site; |
| · | clinical trials of our product candidates may fail to show safety or efficacy or otherwise produce negative or inconclusive results, and we may decide, or regulatory authorities may require us, to conduct additional clinical trials or abandon drug development programs; |
| · | the number of patients required for clinical trials of our product candidates may be larger than we anticipate; |
| · | enrollment in our clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
| · | difficulties in having subjects complete a clinical trial or returning for post-treatment follow-up; |
| · | changes to clinical trial protocols; |
| · | our third-party contractors, including clinical investigators, contract manufacturers and vendors may fail to comply with applicable regulatory requirements, lose their licenses or permits, or otherwise fail, or lose the ability to, meet their contractual obligations to us in a timely manner, or at all; |
| · | we might have to suspend or terminate clinical trials of our product candidates for various reasons, including a finding that the participants are being exposed to unacceptable health risks; |
43
| · | regulatory authorities or IRBs may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements, a finding that our product candidates have undesirable side effects or other unexpected characteristics, or that the participants are being exposed to unacceptable health risks; |
| · | the cost of clinical trials of our product candidates may be greater than we anticipate, and we may lack adequate funding to continue one or more clinical trials; |
| · | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; |
| · | clinical trial sites may deviate from clinical trial protocol or drop out of a clinical trial; and |
| · | occurrence of serious adverse events in trials of the same class of agents conducted by other companies. |
Adverse side effects, properties or other safety risks associated with PMN310, PMN442, PMN267 or any future product candidates could delay or preclude approval, cause us to suspend or discontinue clinical trials, abandon further development, limit the commercial profile of an approved label or result in significant negative consequences following marketing approval, if any.
As is the case with pharmaceuticals generally, it is possible that there may be side effects and adverse events associated with the use of PMN310, PMN442, PMN267 or any future product candidates we may develop. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics as the clinical trials progress to greater exposures and a larger number of patients. Undesirable side effects caused by, or unexpected or unacceptable characteristics associated with, PMN310, PMN442, PMN267 or any future product candidates we may develop, could result in the delay, suspension or termination of clinical trials by us, the FDA or other regulatory authorities, or IRBs for a number of reasons. We may also elect to limit their development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, which may limit the commercial expectations for such product candidate if approved. If we elect or are required to further delay, suspend or terminate any clinical trial of any product candidates we may develop, the commercial prospects of such product candidates will be harmed and our ability to generate drug revenues from any such product candidates will be delayed or eliminated.
It is possible that, as we test our product candidates in clinical trials, or as the use of a product candidate becomes more widespread if it receives regulatory approval, we may identify additional adverse events that were not identified or not considered significant in our earlier trials. If such side effects become later known in development or upon approval, if any, such findings may harm our business, financial condition, results of operations and prospects significantly. If we or others later identify undesirable side effects, a number of potentially significant negative consequences could result, including:
| · | regulatory authorities may withdraw, suspend or limit approval of a product candidate; |
| · | we may be required to recall a drug or change the way such drug is administered to patients; |
| · | regulatory authorities may require additional warnings or statements in the labeling, such as a boxed warning or a contraindication or issue safety alerts, press releases or other communications containing warnings or other safety information about the product candidate, for example, field alerts to physicians and pharmacies; |
| · | regulatory authorities may require us to implement a REMS to ensure that the benefits of the drug outweigh its risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools; |
44
| · | we may be required to change the way a drug is distributed or administered, conduct additional clinical trials or be required to conduct additional post-marketing studies or surveillance; |
| · | we may be subject to regulatory investigations and government enforcement actions; |
| · | we may decide to remove such product candidates from the market; |
| · | we could be sued and held liable for harm caused to patients; |
| · | sales of the drug may decrease significantly or become less competitive; and |
| · | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of a product candidate, if approved, and could significantly harm our business, financial condition, results of operations and prospects.
We may experience delays or difficulties in the enrollment and retention of patients in clinical trials, which could delay or prevent our receipt of necessary regulatory approvals.
Successful and timely completion of clinical trials will require that we enroll a sufficient number of patients. Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population and competition for patients eligible for our clinical trials with competitors which may have ongoing clinical trials for product candidates that are under development to treat the same indications as one or more of our product candidates or approved products for the conditions for which we are developing our product candidates.
Trials may be subject to delays as a result of patient enrollment taking longer than anticipated or patient withdrawal. We may not be able to initiate or continue clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA, EMA or foreign regulatory authorities. We cannot predict how successful we will be at enrolling subjects in future clinical trials. Subject enrollment is affected by other factors including:
| · | the severity and difficulty of diagnosing the disease under investigation; |
| · | the eligibility and exclusion criteria for the trial in question; |
| · | the size of the patient population and process for identifying patients; |
| · | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| · | the design of the trial protocol; |
| · | the perceived risks and benefits of the product candidate in the trial, including relating to cell therapy approaches; |
| · | the availability of competing commercially available therapies and other competing therapeutic candidates’ clinical trials for the disease or condition under investigation; |
| · | the willingness of patients to be enrolled in our clinical trials; |
45
| · | the efforts to facilitate timely enrollment in clinical trials; |
| · | potential disruptions caused by the COVID-19 pandemic, including difficulties in initiating clinical sites, enrolling and retaining participants, diversion of healthcare resources away from clinical trials, travel or quarantine policies that may be implemented, and other factors; |
| · | the patient referral practices of physicians; |
| · | the ability to monitor patients adequately during and after treatment; and |
| · | the proximity and availability of clinical trial sites for prospective patients. |
Our inability to enroll a sufficient number of patients for clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in these clinical trials may result in increased development costs for our product candidates, which would cause the value of our company to decline and limit our ability to obtain additional financing. Furthermore, we expect to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials and we will have limited influence over their performance.
Furthermore, even if we are able to enroll a sufficient number of patients for our clinical trials, we may have difficulty maintaining enrollment of such patients in our clinical trials.
Interim, “top-line” and preliminary results from our clinical trials that we announce or publish from time to time may change as more data become available and is subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim, top-line or preliminary results from our clinical trials. Interim results from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or top-line results also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are reported. Differences between preliminary, top-line or interim data and final data could significantly harm our business prospects and may cause the trading price of our Common Shares to fluctuate significantly. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated.
Further, others, including regulatory agencies may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular development program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure. Any information we determine not to disclose may ultimately be deemed meaningful by you or others with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product candidate or our business. If the interim, top-line or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, product candidates may be harmed, which could significantly harm our business prospects.
46
We cannot be certain that PMN310, PMN442, PMN267 or any of our future product candidates will receive regulatory approval, and without regulatory approval we will not be able to market our product candidates.
We currently have no product candidates approved for sale and we cannot guarantee that we will ever have marketable product candidates. Our ability to generate revenue related to sales of PMN310, PMN442, and PMN267, if ever, will depend on the successful development and regulatory approval of such product candidates.
The development of a product candidate and its approval and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, distribution, import and export are subject to extensive regulation by the FDA, the EMA and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the U.S., Europe or other countries until we receive approval of a BLA from the FDA or MAA from the EMA, respectively. We have not submitted any marketing applications for any product candidate.
BLAs and MAAs, and other foreign equivalents must include extensive nonclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. BLAs and MAAs must also include significant information regarding the chemistry, manufacturing and controls for the drug. Obtaining approval of a BLA or a MAA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA and the EMA review processes can take years to complete and approval is never guaranteed. If we submit a BLA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators of other jurisdictions, such as the EMA, have their own procedures for approval of product candidates.
Even if a drug is approved, the FDA or the EMA, or other foreign equivalent, as the case may be, may limit the indications for which the drug may be marketed, require extensive warnings on the drug labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the U.S. and Europe also have requirements for approval of product candidates with which we must comply prior with marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. In addition, delays in approvals or rejections of marketing applications in the U.S., Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, nonclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of drug development and the emergence of new information regarding PMN310, PMN442, PMN267 or other product candidates we may develop in the future. Also, regulatory approval for any of our product candidates may be withdrawn.
Before we submit a BLA to the FDA or a MAA to the EMA for a product candidate, we will be required to successfully complete our clinical trials. The FDA generally requires two pivotal clinical trials to support approval. In addition, we must scale up manufacturing and complete other standard nonclinical and clinical studies. We cannot predict whether clinical trials will be successful or whether regulators will agree with our conclusions regarding the nonclinical studies and the clinical trials we conduct.
Our lead product candidate, PMN310, is being developed for the treatment of AD, a disease that has seen limited success in drug development.
Efforts by biopharmaceutical and pharmaceutical companies in treating AD have seen limited success in drug development. Only one disease-modifying therapeutic option has been approved by the FDA. Biogen’s Aduhelm, a mAb administered via infusion, received accelerated approval from the FDA on June 7, 2021. We cannot be certain that our oral, small-molecule approach will lead to the development of approvable or marketable products. With the exception of Aduhelm, the only drugs approved by the FDA to treat patients with AD address the symptoms of the disease. Since 2003, over 500 clinical studies have been completed and only Aduhelm has been approved by the FDA. As a result, the FDA has a limited set of products to rely on in evaluating PMN310. This could result in a longer than expected regulatory review process, increased expected development costs or the delay or prevention of commercialization of PMN310 for the treatment of AD.
In addition to the significant uncertainty related to insurance coverage and reimbursement of all newly-approved products, there is greater uncertainty for products approved for the treatment of AD. For example, the yearly wholesale acquisition cost of the maintenance dose of Aduhelm is $28,200. The CMMS recently issued a draft determination that proposes to cover the cost of anti-amyloid monoclonal antibodies, including Aduhelm, only in the context of clinical trials approved by CMMS or by the National Institutes of Health. They include only randomized controlled trials conducted in hospital-based outpatient settings, and require patient diversity reflecting that of the U.S. population diagnosed with AD. CMMS does not presently intend to cover the cost of these drugs outside the context of clinical trials.
We may in the future conduct clinical trials for our product candidates outside the U.S., and the FDA, EMA and other foreign regulatory authorities may not accept data from such trials.
We may in the future choose to conduct one or more of our clinical trials outside the U.S., including in Europe. The acceptance of study data from clinical trials conducted outside the U.S. or another jurisdiction by the FDA, EMA or applicable foreign regulatory authorities may be subject to certain conditions or may not be accepted at all. In cases where data from foreign clinical trials are intended to serve as the basis for marketing approval in the U.S., the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the U.S. population and U.S. medical practice; and (ii) the trials were performed by clinical investigators of recognized competence and pursuant to cGCP, regulations. Additionally, the FDA’s clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory bodies have similar approval requirements. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA, EMA or any other foreign regulatory authority will accept data from trials conducted outside of the U.S. or the applicable jurisdiction. If the FDA, EMA or any applicable foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan, and which may result in our product candidates not receiving approval or clearance for commercialization in the applicable jurisdiction.
47
We may not be successful in our efforts to build a pipeline of additional product candidates.
We may not be able to identify and successfully develop new product candidates. Even if we are successful in building our product pipeline, the potential product candidates that we identify may not be suitable for clinical development or, if deemed suitable for clinical development, successful in any clinical trials. For example, product candidates may be shown to have harmful side effects or other characteristics that indicate that they are unlikely to be successfully developed, much less receive marketing approval and achieve market acceptance. If we do not successfully develop and commercialize product candidates, we will not be able to obtain product revenue in future periods, which would result in significant harm to our financial position and adversely affect our share price.
If we do not achieve our projected development goals in the time frames we announce and expect, the commercialization of our products may be delayed.
From time to time, we may estimate the timing of the accomplishment of various scientific, clinical, regulatory, manufacturing and other product development goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of nonclinical studies and clinical trials and the submission of regulatory filings, including BLA submissions. From time to time, we may publicly announce the expected timing of some of these milestones. All of these milestones are, and will be, based on a variety of assumptions. The actual timing of these milestones can vary significantly compared to our estimates, in some cases for reasons beyond our control. We may experience numerous unforeseen events during, or as a result of, any future clinical trials that we conduct that could delay or prevent our ability to receive marketing approval or commercialize our product candidates.
We may develop PMN310, PMN442, PMN267 and future product candidates for use in combination with other therapies, which could expose us to additional regulatory risks.
We may develop PMN310, PMN442, PMN267 and future product candidates for use in combination with one or more other approved therapies for the disease state being studied. Even if any product candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risk that the FDA, EMA or comparable foreign regulatory authorities could revoke approval of the therapy used in combination with our product candidate or that safety, efficacy, manufacturing or supply issues could arise with these existing therapies. This could result in our own products being removed from the market or being less successful commercially.
Further, we will not be able to market and sell any product candidate we develop in combination with an unapproved therapy for a combination indication if that unapproved therapy does not ultimately obtain marketing approval either alone or in combination with our product. In addition, unapproved therapies face the same risks described with respect to our product candidates currently in development, including the potential for serious adverse effects, delay in their clinical trials and lack of FDA approval.
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through nonclinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and product characteristics.
Such changes carry the risk that they will not achieve our intended objectives. Any such changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes. Such changes may also require additional testing, FDA notification approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence sales and generate revenue. In addition, we may be required to make significant changes to our upstream and downstream processes across our pipeline, which could delay the development of our future product candidates.
48
Risks Related to the Commercialization of Our Product Candidates
Successful commercialization of our products, if approved, will depend on a number of factors and we cannot guarantee that we will be able to successfully commercialize our products.
Successful commercialization of our products, if at all, will depend on a number of factors, including our ability to:
| · | raise sufficient capital to fund future commercialization efforts; |
| · | build a commercial team and supporting organizational infrastructure; |
| · | obtain necessary licenses, on commercially reasonable terms, for certain offerings the Company may contemplate; |
| · | establish partnerships and alliances with third parties to secure commercial capabilities that we may not wish to build; |
| · | market and distribute our products; |
| · | distinguish our products from others available on the market; |
| · | obtain any necessary regulatory approvals for our facilities, products and processes; |
| · | gain reimbursement by third-party payors, such as private health insurers, managed-health organizations, and state-sponsored health insurance plans for each jurisdiction in which our products are offered; |
| · | educate physicians and change physician behavior to secure clinical adoption of our products; |
| · | promote awareness of our products to increase market penetration; and |
| · | publish in peer-reviewed journals. |
There is no assurance that we will be successful in these areas. Any failure or delay in such areas could have a material adverse impact on our business, financial condition, results of operations and prospects.
The market opportunities for PMN310, PMN442, PMN267, and future product candidates, if approved, may be smaller than we anticipate.
We expect to seek approval for product candidates for various neurodegenerative diseases and other mis-folded protein diseases. Our estimates of market potential have been derived from a variety of sources, including scientific literature, patient foundations and market research and may prove to be incorrect. Even if we obtain significant market share for our product candidates after FDA approval, the potential target populations may be too small to consistently generate revenue, and we may never achieve profitability without obtaining marketing approval for additional indications.
49
Even if our current or future product candidates obtain regulatory approval, they may fail to achieve the broad degree of adoption and use by physicians, patients, hospitals, healthcare payors and others in the medical community necessary for commercial success.
Even if one or more of our product candidates receive FDA or other regulatory approvals, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, healthcare payors and others in the medical community. Most of our product candidates target mechanisms for which there are limited or no currently approved products, which may result in slower adoption by physicians, patients and payors. If our product candidates do not achieve an adequate level of acceptance, we may not generate significant product revenue and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
| · | the clinical indications for which the product is approved and patient demand for approved products that treat those indications; |
| · | the safety and efficacy of our product as compared to other available therapies; |
| · | the availability of coverage and adequate reimbursement from governmental healthcare plans or third party payors for any of our product candidates that may be approved; |
| · | acceptance by physicians, operators of clinics and patients of the product as a safe and effective treatment; |
| · | physician and patient willingness to adopt a new therapy over other available therapies to treat approved indications; |
| · | overcoming any biases physicians or patients may have toward particular therapies for the treatment of approved indications; |
| · | proper training and administration of our product candidates by physicians and medical staff; |
| · | public misperception regarding the use of our therapies, if approved for commercial sale; |
| · | patient satisfaction with the results and administration of our product candidates and overall treatment experience, including, for example, the convenience of any dosing regimen; |
| · | the cost of treatment with our product candidates in relation to alternative treatments and reimbursement levels, if any, and willingness to pay for the product, if approved, on the part of insurance companies and other third-party payors, physicians and patients; |
| · | the revenue and profitability that our products may offer a physician as compared to alternative therapies; |
| · | limitations or warnings contained in the FDA-approved labeling for our products; |
| · | any FDA requirement to undertake a REMS; |
| · | the effectiveness of our sales, marketing and distribution efforts; |
| · | adverse publicity about our products or favorable publicity about competitive products; and |
| · | potential product liability claims. |
50
We cannot assure you that our current or future product candidates, if approved, will achieve broad market acceptance among physicians, patients, healthcare payors and others in the medical community. Even if we receive regulatory approval to market any of our product candidates, we cannot assure you that any such product candidate will be more effective than other commercially available alternatives or successfully commercialized. Any approval we may obtain could be for indications or patient populations that are not as broad as intended or desired or may require labeling that includes significant use or distribution restrictions or safety warnings. We may also be required to perform additional or unanticipated clinical trials to obtain approval or be subject to additional post-marketing testing requirements to maintain approval. In addition, regulatory authorities may withdraw their approval of a product or impose restrictions on its distribution, such as in the form of a REMS. Any failure by our product candidates that obtain regulatory approval to achieve market acceptance or commercial success would adversely affect our reputation, ability to raise additional capital, financial condition, results of operations and business prospects.
Our product candidates have never been manufactured on a commercial scale, and there are risks associated with scaling up manufacturing to commercial scale. In particular, we will need to develop a larger scale manufacturing process that is more efficient and cost-effective to commercialize our potential products, which may not be successful.
Our product candidates have never been manufactured on a commercial scale, and there are risks associated with scaling up manufacturing to commercial scale including, among others, cost overruns, potential problems with process scale-up, process reproducibility, stability issues, lot consistency and timely availability of raw materials. There is no assurance that our third-party manufacturers will be successful in establishing a larger-scale commercial manufacturing process for our product candidates which achieves our objectives for manufacturing capacity and cost of goods. In addition, there is no assurance that any third-party manufacturers will be able to manufacture our product candidates to specifications acceptable to the FDA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of such products or to meet potential future demand. Our failure to properly or adequately scale up manufacturing for commercial scale would adversely affect our business, results of operations and financial condition.
The successful commercialization of our product candidates will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage, reimbursement levels and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those drugs and decrease our ability to generate revenue.
The availability and adequacy of coverage and reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and other third-party payors are essential for most patients to be able to afford prescription medications such as our product candidates, if approved. Our ability to achieve acceptable levels of coverage and reimbursement for products by governmental authorities, private health insurers and other organizations will have an effect on our ability to successfully commercialize our product candidates. Even if we obtain coverage for our product candidates by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the U.S., the European Union or elsewhere will be available for our product candidates or any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
Third-party payors increasingly are challenging prices charged for biopharmaceutical products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drugs or biologics when an equivalent generic drug, biosimilar or a less expensive therapy is available. It is possible that a third-party payor may consider our product candidates as substitutable and only offer to reimburse patients for the cost of the less expensive product. Even if we show improved efficacy or improved convenience of administration with our product candidates, pricing of existing third-party therapeutics may limit the amounts we will be able to charge for our product candidates. These payors may deny or revoke the reimbursement status of a given product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in our product candidates. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates and may not be able to obtain a satisfactory financial return on our investment in the development of product candidates.
51
There is significant uncertainty related to the insurance coverage and reimbursement of newly-approved products. In the U.S., third-party payors, and governmental healthcare plans, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs and biologics will be covered. The Medicare and Medicaid programs increasingly are used as models in the U.S. for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers who use such therapies. We cannot predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
No uniform policy for coverage and reimbursement for products exists among third-party payors in the U.S. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases on short notice, and we believe that changes in these rules and regulations are likely.
Outside the U.S., international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe and other foreign jurisdictions have and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amounts that we are able to charge for our product candidates. Accordingly, in markets outside the U.S., the reimbursement for our product candidates may be reduced compared with the U.S. and may be insufficient to generate commercially-reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors in the U.S. and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products, and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of our product candidates due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and biologics and surgical procedures and other treatments, has become intense. As a result, increasingly high barriers are being erected to the entry of new products.
We currently have no sales organization. If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our product candidates effectively in the U.S. and foreign jurisdictions, if approved, or generate product revenue.
We currently do not have a marketing or sales organization. In order to commercialize our product candidates in the U.S. and foreign jurisdictions, if approved, we intend to make arrangements with third parties to perform these services, and we may not be successful in doing so. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our product candidates, if approved. If we are not successful in commercializing our product candidates or any future product candidates, if approved, either on our own or through arrangements with third parties, we may not be able to generate any product revenue and we would incur significant additional losses.
52
Risks Related to Our Financial Position and Capital Needs
We will require additional financing to achieve our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our development programs, commercialization efforts or other operations.
The development of biopharmaceutical therapeutic candidates is capital-intensive. We expect our expenses to increase in connection with our ongoing activities, particularly as we conduct our ongoing and planned preclinical studies of our development programs, initiate clinical trials for our therapeutic candidates and seek regulatory approval for our current therapeutic candidates and any future therapeutic candidates we may develop. If we obtain regulatory approval for any of our therapeutic candidates, we also expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. Because the outcome of any preclinical study or clinical trial is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our therapeutic candidates. Furthermore, following the effectiveness of this Registration Statement, we expect to incur additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. We had working capital of $16.8 million as of December 31, 2021. Although we expect available funds will be sufficient to fund our operating expenses through the end of 2023, we will require substantial additional funds for further research and development, planned clinical testing, regulatory approvals, establishment of manufacturing capabilities and, if necessary, the marketing and sale of our products. Our ability to raise additional financing and maintain operations in the future could be at substantial risk and there can be no assurance that additional funding or partnerships will be available on acceptable terms that would foster successful commercialization of our products. Failing to raise capital when needed or on attractive terms could force us to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
We may attempt to raise additional funds for these purposes through public or private equity or debt financing, collaborations with other biopharmaceutical companies and/or from other sources.
We have no product candidates approved for commercial sale, we have never generated any revenue from sales and we may never be profitable.
We have no product candidates approved for sale, have never generated any revenue from sales, have never been profitable and do not expect to be profitable in the foreseeable future. To date, we have not recorded any revenues from the sale of biopharmaceutical products. As at December 31, 2021, we had a deficit of $62.2 million. The cumulative deficit incurred from when we changed our name and focus in July 2015, through December 31, 2021 was $32.2 million. We expect to incur additional losses during the periods of research and development, clinical testing, and application for regulatory approval of its product candidates. We also expect to incur losses unless and until such time as payments from corporate collaborations, product sales and/or royalty payments generate sufficient revenues to fund its continuing operations.
To date, we have devoted most of our financial resources to research and development of PMN310, including our nonclinical development activities of PMN310, and corporate overhead. We expect that it will be several years, if ever, before we have a product candidate approved and ready for commercialization. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, PMN310 and any other product candidate we may develop in the future, prepare for and begin the commercialization of any approved product candidates and add infrastructure and personnel to support our drug development efforts and operations as a public company. We anticipate that any such losses could be significant for the next several years. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our shareholders’ equity and working capital. Further, these net losses may fluctuate significantly from quarter-to-quarter or year-to-year. To become and remain profitable, we must develop and eventually commercialize PMN310 or another drug with significant revenue.
53
We may never succeed in developing a commercial drug and, even if we succeed in commercializing one or more product candidates, we may never generate revenues that are large enough to achieve profitability. In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown challenges. Because of these numerous risks and uncertainties, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to generate revenues or achieve profitability. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis, and we will continue to incur substantial research and development costs and other expenditures to develop and market additional product candidates.
Risks Related to Our Dependence on Third Parties
We will rely on third parties to supply components, research, develop, test, and manufacture our product candidates and market, if approved. The loss of any of these third party relationships or the failure of any of them to meet their obligations to us could affect our ability to develop and obtain approval of our product candidates in a timely manner.
Our activities will require us to enter into various arrangements with corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, manufacturing, marketing and commercialization of our products. We intend to attract corporate partners and enter into additional research collaborations. There can be no assurance, however, that we will be able to establish such additional collaborations on favorable terms, if at all, or that our current or future collaborations will be successful. Failure to attract commercial partners for our products may result in substantial clinical testing, manufacturing and commercialization costs prior to realizing any revenue from product sales or result in delays or program discontinuance if funds are not available in sufficient quantities.
Should any collaborative partner fail to develop, manufacture, or successfully commercialize any product to which we have rights, or any partner’s product to which we will have rights, our business may be adversely affected. Failure of a collaborative partner to continue to participate in any particular program could delay or halt the development or commercialization of products generated from such program. In addition, there can be no assurance that the collaborative partners will not pursue other technologies or develop alternative products either alone or in collaboration with others, including our competitors, as a means for developing treatments for the diseases targeted by our programs.
Furthermore, we will hold licenses for certain technologies and there can be no assurance that these licenses will not be terminated, or that they will be renewed on acceptable conditions. We intend to negotiate additional licenses in respect of technologies developed by other companies and academic institutions. Terms of license agreements to be negotiated may include, inter alia, a requirement to make milestone payments, which may be substantial. We will also be obligated to make royalty payments on the sales, if any, of products resulting from licensed technology and, in some instances, may be responsible for the costs of filing and prosecuting patent applications.
54
We intend to rely on contract research organizations, or CROs, and other third parties to conduct, supervise and monitor a significant portion of our research and nonclinical testing and clinical trials for our product candidates, and if those third parties do not successfully carry out their contractual duties, comply with regulatory requirements or otherwise perform satisfactorily, we may not be able to obtain regulatory approval or commercialize product candidates, or such approval or commercialization may be delayed, and our business may be substantially harmed.
We intend to engage CROs and other third parties to conduct our planned nonclinical studies or clinical trials, and to monitor and manage data. We expect to continue to rely on third parties, including clinical data management organizations, medical institutions and clinical investigators, in the future. Any of these third parties may terminate their engagements with us, some in the event of an uncured material breach and some at any time for convenience. If any of our relationships with these third parties terminate, we may not be able to timely enter into arrangements with alternative third parties or to do so on commercially reasonable terms, if at all. Switching or adding CROs involves substantial cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. Further, the performance of our CROs and other third parties conducting our trials may also be interrupted by the ongoing COVID-19 pandemic, including due to travel or quarantine policies, heightened exposure of a CRO or clinical site or other vendor staff who are healthcare providers to COVID-19 or prioritization of resources toward the pandemic.
In addition, any third parties conducting our clinical trials will not be our employees, and, except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our clinical programs. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. Consequently, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase substantially and our ability to generate revenue could be delayed significantly.
We rely on these parties for execution of our nonclinical studies and clinical trials and generally do not control their activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with Good Clinical Practices, or GCPs, which are standards for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. If we or any of our CROs or other third parties, including trial sites, fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMPs conditions. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process, or may result in fines, adverse publicity and civil and criminal sanctions.
We also are required to register certain ongoing clinical trials and post the results of certain completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval for PMN310 or any other product candidate we develop.
55
We also expect to rely on other third parties to store and distribute product supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential revenue.
If any of our third-party manufacturers encounter difficulties in production of PMN310, PMN442, PMN267 or any future product candidate we develop, or fail to meet rigorously enforced regulatory standards, our ability to provide supply of our product candidates for clinical trials or, if approved, for commercial sale could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure.
The processes involved in manufacturing PMN310, PMN442, PMN267 and any other product candidate we may develop are highly-regulated and subject to multiple risks. As product candidates are developed through nonclinical studies to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials. When changes are made to the manufacturing process, we may be required to provide preclinical and clinical data showing the comparable identity, strength, quality, purity or potency of the products before and after such changes. If microbial, viral or other contaminations are discovered at the facilities of our third-party manufacturers, such facilities may need to be closed for an extended period of time to investigate and remedy the contamination, which could delay clinical trials and adversely harm our business.
In order to conduct clinical trials of our product candidates, or supply commercial product candidates, if approved, we will need to manufacture them in both small and large quantities. We currently rely on third parties to manufacture our product candidates, and our manufacturing partners will have to modify and scale-up the manufacturing process when we transition to commercialization of our product candidates, if approved. Our manufacturing partners may be unable to successfully modify or scale-up the manufacturing capacity for any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If our manufacturing partners are unable to successfully scale-up the manufacture of our product candidates in sufficient quality and quantity, the development, testing and clinical trials of that product candidate may be delayed or become infeasible, and regulatory approval or commercial launch of any resulting product may be delayed or not obtained, which could significantly harm our business. In addition, building internal manufacturing capacity would carry significant risks in terms of being able to plan, design and execute on a complex project to build manufacturing facilities in a timely and cost-efficient manner.
In addition, the manufacturing process for any product candidates that we may develop will be subject to FDA, EMA and foreign regulatory requirements, and continuous oversight, and we will need to contract with manufacturers who can meet all applicable FDA, EMA and foreign regulatory authority requirements, including complying with cGMPs on an ongoing basis. If we or our third-party manufacturers are unable to reliably produce product candidates in accordance with the requirements of the FDA, EMA or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such product candidates. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our third party contract manufacturers will be able to manufacture the approved product in accordance with the requirements of the FDA, EMA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates, impair commercialization efforts, increase our cost of goods and have an adverse effect on our business, financial condition, results of operations and growth prospects. Significant non-compliance could also result in the imposition of sanctions, including warning or untitled letters, fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approvals for our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could damage our reputation and our business.
56
We will likely seek collaborations with third parties for the development or commercialization of our product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of those product candidates, including PMN310, PMN442, and PMN267.
We will likely seek third-party collaborators for the commercialization of PMN310, PMN442, and PMN267 and any of our future product candidates, in the U.S. and may enter into collaboration agreements for the development and commercialization of any of our product candidates outside the U.S. In the U.S., commercialization partners are likely to include large biotechnology or pharmaceutical companies. Our likely collaborators outside the U.S. would most likely include regional and national pharmaceutical companies and biotechnology companies. If we enter into such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenue from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates would pose the following risks to us:
| · | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| · | collaborators may not perform their obligations as expected; |
| · | collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities; |
| · | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| · | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| · | we could grant exclusive rights to our collaborators that would prevent us from collaborating with others; |
| · | product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or drugs, which may cause collaborators to cease to devote resources to the commercialization of our product candidates; |
| · | a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such products; |
| · | disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive; |
57
| · | collaborators may not properly maintain or defend our or their intellectual property rights or may use our or their proprietary information in such a way as to invite litigation that could jeopardize or invalidate such intellectual property or proprietary information or expose us to potential litigation; |
| · | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and |
| · | collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If any future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for any collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, EMA or similar regulatory authorities outside the U.S., the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate additional collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of such product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate revenue.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain sufficient intellectual property protection for our product candidates, and other proprietary technologies we develop, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our product candidates, and other proprietary technologies if approved, may be adversely affected.
Our commercial success will depend in part on our ability to obtain and maintain a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our product candidates, and other proprietary technologies we develop. If we are unable to obtain or maintain patent protection with respect to our product candidates, and other proprietary technologies we may develop, our business, financial condition, results of operations, and prospects could be materially harmed.
58
The patent position of biotechnology and pharmaceutical companies is highly uncertain and involves complex legal, scientific, and factual questions and has been the subject of frequent litigation in recent years. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Our patent applications may not result in patents being issued that protect our product candidates and other proprietary technologies we may develop or that effectively prevent others from commercializing competitive technologies and products. Further, no consistent policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the U.S. or in many jurisdictions outside of the U.S. Changes in either the patent laws or interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that may be issued from the applications we may own or license from third parties. Further, if any patents we obtain or license are deemed invalid and unenforceable, our ability to commercialize or license our technology could be adversely affected.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or any of our actual or potential future collaborators will be successful in protecting our product candidates and other proprietary technologies and their uses by obtaining, defending and enforcing patents. These risks and uncertainties include the following:
| · | the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process, the noncompliance with which can result in abandonment or lapse of a patent or patent application, and partial or complete loss of patent rights in the relevant jurisdiction; |
| · | patent applications may not result in any patents being issued; |
| · | issued patents may be challenged, invalidated, modified, revoked, circumvented, found to be unenforceable, or may otherwise not provide any competitive advantage; |
| · | our competitors, many of whom have substantially greater resources than we do and many of whom have made significant investments in competing technologies, may seek or may have already obtained patents that will limit, interfere with, or eliminate our ability to make, use and sell our product candidates; |
| · | other parties may have designed around our claims or developed technologies that may be related or competitive to ours, may have filed or may file patent applications and may have received or may receive patents that overlap or conflict with our patent applications and/or patents, either by claiming the same composition of matter, methods or formulations or by claiming subject matter that could dominate our patent position; |
| · | any successful opposition to any patents owned by or licensed to us could deprive us of rights necessary for the practice of our technologies or the successful commercialization of any product candidate that we may develop; |
| · | because patent applications in the U.S. and most other countries are confidential for a period of time after filing, we cannot be certain that we or our licensors were the first to file any patent application related to our product candidates and other proprietary technologies and their uses; |
| · | an interference proceeding can be provoked by a third party or instituted by the USPTO to determine who was the first to invent any of the subject matter covered by the patent claims of any application with an effective filing date before March 16, 2013; |
59
| · | there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the U.S. for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; and |
| · | countries other than the U.S. may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop, and market competing product candidates in those countries. |
The patent prosecution process is expensive, time-consuming, and complex, and we may not be able to file, prosecute, or maintain all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Although we enter into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors and other third parties, any of these parties may breach such agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection for such output. In addition, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our inventions and the prior art allow our inventions to be patentable over the prior art. Furthermore, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. If we do not adequately protect our intellectual property and proprietary technology, competitors may be able to use our product candidates and other proprietary technologies and erode or negate any competitive advantage we may have, which could have a material adverse effect on our financial condition and results of operations. For example:
| · | others may be able to make compounds that are similar to our product candidates but that are not covered by the claims of our patents; |
| · | we might not have been the first to file patent applications for these inventions; |
| · | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| · | any patents that we obtain may not provide us with any competitive advantages; |
| · | we may not develop additional proprietary technologies that are patentable; |
| · | our competitors might conduct research and development activities in countries where we do not have patent rights or where patent protection is weak and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| · | we cannot ensure that we will be able to successfully commercialize our product candidates on a substantial scale, if approved, before the relevant patents that we own or license expire; or |
| · | the patents of others may have an adverse effect on our business. |
Others have filed, and in the future are likely to file, patent applications covering products and technologies that are similar, identical or competitive to ours or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed or in-licensed by us, or that we or our licensors will not be involved in interference, opposition or invalidity proceedings before U.S. or non-U.S. patent offices.
60
We cannot be certain that claims in an issued patent covering our product candidates will be considered patentable by the USPTO, courts in the U.S., or by patent offices and courts in foreign countries. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the U.S. As a result, we may encounter significant problems in protecting and defending our intellectual property internationally.
The strength of patents in the biotechnology and pharmaceutical fields involves complex legal and scientific questions and can be uncertain. Patent applications that we file or in-license may fail to result in issued patents with claims that cover our product candidates in the U.S. or in foreign countries. Even if such patents do successfully issue, third parties may challenge the ownership, validity, enforceability, or scope thereof, which may result in such patents being narrowed, invalidated, or held unenforceable. Any successful opposition to our patents could deprive us of exclusive rights necessary for the successful commercialization of our product candidates. Furthermore, even if they are unchallenged, our patents may not adequately protect our intellectual property, provide exclusivity for our product candidates or prevent others from designing around our claims. If the breadth or strength of protection provided by our patents with respect to our product candidates is threatened, it could dissuade companies from collaborating with us to develop, or threaten our ability to commercialize our product candidates.
For U.S. patent applications in which claims are entitled to a priority date before March 16, 2013, an interference proceeding can be provoked by a third party or instituted by the USPTO to determine who was the first to invent any of the subject matter covered by the patent claims of our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our participation in an interference proceeding may fail and, even if successful, may result in substantial costs and distract our management and other employees.
For U.S. patent applications containing a claim not entitled to priority before March 16, 2013, there is greater level of uncertainty in the patent law. In September 2011, the Leahy-Smith America Invents Act, or America Invents Act, was signed into law. The America Invents Act includes a number of significant changes to U.S. patent law, including provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The USPTO is developing regulations and procedures to govern the administration of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and in particular, the “first to file” provisions, were enacted on March 16, 2013. This will require us to be cognizant going forward of the time from invention to filing of a patent application and be diligent in filing patent applications, but circumstances could prevent us from promptly filing patent applications on our inventions. It remains unclear what impact the America Invents Act will have on the operation of our business. As such, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
The term of any individual patent depends on applicable law in the country where the patent is granted. In the U.S., provided all maintenance fees are timely paid, a patent generally has a term of 20 years from its application filing date or earliest claimed non-provisional filing date. Extensions may be available under certain circumstances, but the life of a patent and, correspondingly, the protection it affords is limited. When the terms of all patents covering our product candidates expire, our business may become subject to competition from competitive products, including biosimilar version of our products.
61
Our product candidates are protected by certain patents, which expire at varying times. We cannot be certain that we will file and, if filed, obtain patent protection for our product candidates beyond our rights in our current patent portfolio. If we are unable to obtain additional patent protection on our product candidates, our primary protection from biosimilar market entries will be limited to regulatory biologic exclusivity.
If we do not obtain patent term extension for our product candidates our business may be materially harmed.
Depending upon the timing, duration, and specifics of any FDA marketing approval of our product candidates, one or more of patents issuing from U.S. patent applications that we file or license may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term, or PTE, of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. Similar patent term restoration provisions to compensate for commercialization delay caused by regulatory review are also available in certain foreign jurisdictions, such as in Europe under Supplemental Protection Certificate, or SPC. If we encounter delays in our development efforts, including our future clinical trials, the period of time during which we could market our product candidates under patent protection would be reduced. Additionally, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents, or otherwise fail to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration, or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product will be shortened and our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
Licensing of intellectual property rights is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise between us and our licensors regarding intellectual property rights subject to a license agreement, including:
| · | the scope of rights granted under the license agreement and other interpretation-related issues; |
| · | whether and the extent to which our technology and processes infringe on intellectual property rights of the licensor that are not subject to the license agreement; |
| · | our right to sublicense intellectual property rights to third parties under collaborative development relationships; |
| · | our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations; and |
| · | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners. |
If disputes over intellectual property rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms and/or to secure our rights to the licensed intellectual property, our business, results of operations, financial condition, and prospects may be adversely affected. We may enter into additional licenses in the future and if we fail to comply with obligations under those agreements, we could suffer adverse consequences.
62
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions during the patent process. Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on any issued patents and/or applications are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ outside counsel to pay these fees due to foreign patent agencies. While an inadvertent lapse may sometimes be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market with similar or identical products or technology earlier than should otherwise have been the case, which would have a material adverse effect on our business, financial condition, results of operations, and prospects.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property, particularly on obtaining and enforcing patents. Our patent rights may be affected by developments or uncertainty in U.S. or foreign patent statutes, patent case law, USPTO rules and regulations or the rules and regulations of foreign patent offices. Obtaining and enforcing patents in the biotechnology and pharmaceutical industry involve both technological and legal complexity, and is therefore costly, time-consuming and inherently uncertain. In addition, the U.S. may, at any time, enact changes to U.S. patent law and regulations, including by legislation, by regulatory rule-making, or by judicial precedent, that adversely affect the scope of patent protection available and weaken the rights of patent owners to obtain patents, enforce patent infringement and obtain injunctions and/or damages. For example, the scope of patentable subject matter under 35 U.S.C. 101 has evolved significantly over the past several years as the Court of Appeals for the Federal Circuit and the Supreme Court issued various opinions, and the USPTO modified its guidance for practitioners on multiple occasions. Other countries may likewise enact changes to their patent laws in ways that adversely diminish the scope of patent protection and weaken the rights of patent owners to obtain patents, enforce patent infringement, and obtain injunctions and/or damages.
Further, the U.S. and other governments may, at any time, enact changes to law and regulation that create new avenues for challenging the validity of issued patents. For example, the America Invents Act created new administrative post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings that allow third parties to challenge the validity of issued patents. This applies to all of our U.S. patents, even those issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
63
We may not be able to protect our intellectual property rights throughout the world.
Patents are of national or regional effect. Filing, prosecuting, and defending patents on our product candidates, and other proprietary technologies we develop in all countries throughout the world would be prohibitively expensive. In addition, the laws of some foreign countries do not protect intellectual property rights in the same manner and to the same extent as laws in the U.S. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement of such patent protection is not as strong as that in the U.S. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
The requirements for patentability may differ in certain countries. For example, unlike other countries, China has a heightened requirement for patentability, and specifically requires a detailed description of medical uses of a claimed drug. In India, unlike the U.S., there is no link between regulatory approval for a drug and its patent status. In addition to India, certain countries in Europe and developing countries, including China, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors.
In those countries, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology or pharmaceutical products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We may become subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees (including former employees of our licensors), collaborators or other third parties have an interest in our patents rights, trade secrets, or other intellectual property as an inventor or co-inventor. The failure to name the proper inventors on a patent application can result in the patents issuing thereon being unenforceable. For example, we may have inventorship disputes arise from conflicting views regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign nationals are involved in the development of the subject matter of the patent, conflicting obligations of third parties involved in developing our product candidates or as a result of questions regarding co-ownership of potential joint inventions. Litigation may be necessary to resolve these and other claims challenging inventorship and/or ownership. Alternatively, or additionally, we may enter into agreements to clarify the scope of our rights in such intellectual property. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business, financial condition, results of operations and prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
64
We may not be successful in obtaining or maintaining necessary rights to product components and processes for our development pipeline through in-licenses.
Presently we have intellectual property rights to our product candidates through a license from the UBC. We also have an intellectual property license through a license with UHN, and, if this agreement remains in place, we could be required to pay a low single digit royalty on revenues to UHN and a low to high single digit royalty on revenues to UBC in the future. Because our program may require the use of additional proprietary rights held by third parties, the growth of our business will likely depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our product candidates may require specific formulations to work effectively and efficiently and these rights may be held by others. We may be unable to acquire or in-license, on reasonable terms, proprietary rights related to any compositions, formulations, methods of use, processes or other intellectual property rights from third parties that we identify as being necessary for our product candidates. Even if we are able to obtain a license to such proprietary rights, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology.
Where we obtain licenses from or collaborate with third parties, we may not have the right to control the preparation, filing, and prosecution of patent applications, or to maintain or enforce the patents, covering technology that we license from third parties, or such activities, if controlled by us, may require the input of such third parties. If any of our licensors or collaboration partners fail to prosecute, maintain and enforce such patents and patent applications in a manner consistent with the best interests of our business, including by payment of all applicable fees for patents covering our product candidates, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products. In addition, even where we have the right to control patent prosecution of patents and patent applications we have licensed from third parties, we may still be adversely affected or prejudiced by actions or inactions of our licensors and their counsel that took place prior to the date upon which we assumed control over patent prosecution. We may also require the cooperation of our licensors and collaborators to enforce any licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business, or in compliance with applicable laws and regulations, which may affect the validity and enforceability of such patents or any patents that may issue from such application.
Moreover, we will likely have obligations under our current or future licenses, including making royalty and milestone payments, and any failure to satisfy those obligations could give our licensor the right to terminate the license. Termination of a necessary license, or expiration of licensed patents or patent applications, could have a material adverse impact on our business. Our business would suffer if any such licenses terminate, if the licensors fail to abide by the terms of the license, if the licensors fail to enforce licensed patents against infringing third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms. Furthermore, if any licenses terminate, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties may gain the freedom to seek regulatory approval of, and to market, products identical or similar to ours. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize products, we may be unable to achieve or maintain profitability.
The licensing and acquisition of third-party proprietary rights is a competitive area, and companies, which may be more established, or have greater resources than we do, may also be pursuing strategies to license or acquire third-party proprietary rights that we may consider necessary or attractive in order to commercialize our product candidates. More established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
65
For example, we have collaborated and may in the future collaborate with U.S. and foreign academic institutions to accelerate our preclinical research or development under written agreements with these institutions. Typically, these institutions provide us with an option to negotiate an exclusive license to any of the institution’s proprietary rights in technology resulting from the collaboration. Regardless of such option to negotiate a license, we may be unable to negotiate a license within the specified time frame or under terms that are acceptable to us. If we are unable to do so, the institution may offer, on an exclusive basis, their proprietary rights to other parties, potentially blocking our ability to pursue our program. In addition, disputes may arise under our existing or future license agreements with these institutions or with other counterparties which may, among other things, lead to the termination or renegotiation of these agreements, or otherwise require us to incur significant financial obligations.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us, either on reasonable terms, or at all. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment, or at all. If we are unable to successfully obtain rights to required third-party intellectual property rights on commercially reasonable terms, our ability to commercialize our products, and our business, financial condition, and prospects for growth, could suffer.
Third-party claims alleging intellectual property infringement may prevent or delay our drug discovery and development efforts.
Our success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the U.S., involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including inter partes review, interference and reexamination proceedings before the USPTO, or oppositions and other comparable proceedings in foreign jurisdictions. The America Invents Act introduced new procedures including inter partes review and post grant review. The implementation of these procedures brings uncertainty to the possibility of challenges to our patents in the future and the outcome of such challenges. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our activities related to our product candidates may give rise to claims of infringement of the patent rights of others.
The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. We cannot assure you that any of our current or future product candidates will not infringe existing or future patents. We may not be aware of patents that have already issued that a third party might assert are infringed by one of our current or future product candidates.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware with claims to materials, compositions, formulations, methods of manufacture or methods for treatment related to our product candidates, or the use or manufacture of our product candidates. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be currently pending third-party patent applications which may later result in issued patents that our product candidates, and other proprietary technologies may infringe, or which such third parties claim are infringed by the use of our technologies. Parties making claims against us for infringement or misappropriation of their intellectual property rights may seek and obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize our product candidates. Defense of these claims, regardless of their merit, could involve substantial expenses and could be a substantial diversion of management and other employee resources from our business.
66
If we collaborate with third parties in the development of technology in the future, our collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to litigation or potential liability. Further, collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability. In the future, we may agree to indemnify our commercial collaborators against certain intellectual property infringement claims brought by third parties.
Any claims of patent infringement asserted by third parties would be time-consuming and could:
| · | result in costly litigation; |
| · | divert the time and attention of our technical personnel and management; |
| · | cause development delays; |
| · | prevent us from commercializing our product candidates until the asserted patent expires or is finally held invalid, unenforceable, or not infringed in a court of law; |
| · | require us to develop non-infringing technology, which may not be possible on a cost-effective basis; |
| · | require us to pay damages to the party whose intellectual property rights we may be found to be infringing, which may include treble damages if we are found to have been willfully infringing such intellectual property; |
| · | require us to pay the attorney’s fees and costs of litigation to the party whose intellectual property rights we may be found to be willfully infringing; and/or |
| · | require us to enter into royalty or license agreements, which may not be available on commercially reasonable terms, or at all. |
If we are sued for patent infringement, we would need to demonstrate that our products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do either. Proving invalidity or unenforceability is difficult. For example, in the U.S., proving invalidity before federal courts requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and divert management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, which may not be available, defend an infringement action or challenge the validity or enforceability of the patents in court. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid or unenforceable, we may incur substantial monetary damages, encounter significant delays in bringing our product candidates to market and be precluded from developing, manufacturing or selling our product candidates.
We do not always conduct independent reviews of pending patent applications of and patents issued to third parties. We cannot be certain that any of our or our licensors’ patent searches or analyses, including but not limited to the identification of relevant patents, analysis of the scope of relevant patent claims or determination of the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending application in the U.S., Europe and elsewhere that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction, because:
| · | some patent applications in the U.S. may be maintained in secrecy until the patents are issued; |
| · | patent applications in the U.S. and elsewhere can be pending for many years before issuance, or unintentionally abandoned patents or applications can be revived; |
67
| · | pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our technologies, our product candidates or their uses; |
| · | identification of third-party patent rights that may be relevant to our technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases, and the difficulty in assessing the meaning of patent claims; |
| · | patent applications in the U.S. are typically not published until 18 months after the priority date; and |
| · | publications in the scientific literature often lag behind actual discoveries. |
Furthermore, the scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history and can involve other factors such as expert opinion. Our interpretation of the relevance or the scope of claims in a patent or a pending application may be incorrect, which may negatively impact our ability to market our products. Further, we may incorrectly determine that our technologies or product candidates are not covered by a third party patent or may incorrectly predict whether a third party’s pending patent application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the U.S. or internationally that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our products or product candidates.
Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours, and others may have or obtain patents or proprietary rights that could limit our ability to make, use, sell, offer for sale or import our product candidates or future products or impair our competitive position. Numerous third-party U.S. and foreign issued patents and pending patent applications exist in the fields in which we are developing product candidates. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Any such patent application may have priority over one of our patent applications, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the U.S. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions. Other countries have similar laws that permit secrecy of patent applications and may be entitled to priority over our applications in such jurisdictions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
If a third party prevails in a patent infringement lawsuit against us, we may have to stop making and selling the infringing product, pay substantial damages, including treble damages and attorneys’ fees if we are found to be willfully infringing a third party’s patents, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure.
We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize our product candidates, which could harm our business significantly. Even if we were able to obtain a license, the rights may be nonexclusive, which may give our competitors access to the same intellectual property.
68
We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industries, in addition to our employees, we engage the services of consultants to assist us in the development of our product candidates, and other proprietary technologies. Many of these consultants, and many of our employees, were previously employed at, or may have previously provided or may be currently providing consulting services to, other pharmaceutical companies including our competitors or potential competitors. We may become subject to claims that we, our employees or a consultant inadvertently or otherwise used or disclosed trade secrets or other information proprietary to their former employers or their former or current clients. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely affect our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to our management team and other employees.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming, and unsuccessful. Further, our issued patents could be found invalid or unenforceable if challenged in court, and we may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
Third parties including competitors may infringe, misappropriate or otherwise violate our patents, patents that may issue to us in the future, or the patents of our licensors that are licensed to us. To counter infringement or unauthorized use, we may need to or choose to file infringement claims, which can be expensive and time-consuming. We may not be able to prevent, alone or with our licensors, infringement, misappropriation, or other violation of our intellectual property, particularly in countries where the laws may not protect those rights as fully as in the U.S., or if we require, but do not receive, the consent or cooperation of our licensors to enforce such intellectual property.
If we choose to go to court to stop another party from using the inventions claimed in our patents, that individual or company has the right to ask the court to rule that such patents are invalid, unenforceable, or should not be enforced against that third party for any number of reasons. In patent litigation in the U.S., defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include an alleged failure to meet any of several statutory requirements for patentability, including lack of novelty, obviousness, lack of written description, indefiniteness, or non-enablement. Grounds for an unenforceability assertion could include an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution, i.e., committed inequitable conduct. Third parties may also raise similar claims before the USPTO, even outside the context of litigation. Similar mechanisms for challenging the validity and enforceability of a patent exist in foreign patent offices and courts and may result in the revocation, cancellation, or amendment of any foreign patents we or our licensors hold now or in the future. The outcome following legal assertions of invalidity and unenforceability is unpredictable, and prior art could render our patents or those of our licensors invalid. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on such product candidate. Such a loss of patent protection would have a material adverse impact on our business.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to conduct our future clinical trials, continue our research programs, license necessary technology from third parties, or enter into development or manufacturing partnerships that would help us bring our product candidates to market.
69
We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace. Even if resolved in our favor, litigation or other legal proceedings relating to our intellectual property rights may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our Common Shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Our ability to enforce our patent rights depends on our ability to establish standing in a court of competent jurisdiction. Whether a patent holder or licensee of a patent has standing can be uncertain and the considerations complex. However, if a licensor is required to be joined, and they are unwilling to do so, we may be unable to proceed with an infringement action.
Our ability to enforce our patent rights depends on our ability to detect infringement. It may be difficult to detect infringers who do not advertise the components or methods that are used in connection with their products and services. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product or service. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
Because of the expense and uncertainty of litigation, we may not be in a position to enforce our intellectual property rights against third parties.
Because of the expense and uncertainty of litigation, we may conclude that even if a third party is infringing our issued patent or patents that may issue from patent applications or other intellectual property rights, the risk-adjusted cost of bringing and enforcing such a claim or action may be too high or not in the best interest of our company or our shareholders. In such cases, we may decide that the more prudent course of action is to simply monitor the situation or initiate or seek some other non-litigious action or solution.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and/or other advisors, and inventions agreements with employees, consultants, and advisors, to protect our trade secrets and other proprietary information. In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly accepted physical and technological security measures. Despite these efforts, we cannot provide any assurances that all such agreements have been duly executed, and these agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
70
In addition, such security measures may not provide adequate protection for our proprietary information, for example, in the case of misappropriation of a trade secret by an employee, consultant, customer, or third party with authorized access. Our security measures may not prevent an employee, consultant or customer from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Monitoring unauthorized uses and disclosures is difficult, and we do not know whether the steps we have taken to protect our proprietary technologies will be effective. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Even though we use commonly accepted security measures, the criteria for protection of trade secrets can vary among different jurisdictions.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. Moreover, third parties may still obtain this information or may come upon this or similar information independently, and we would have no right to prevent them from using that technology or information to compete with us. Trade secrets will over time be disseminated within the industry through independent development, the publication of journal articles, and the movement of personnel skilled in the art from company to company or academic to industry scientific positions. Though our agreements with third parties typically restrict the ability of our advisors, employees, collaborators, licensors, suppliers, third-party contractors, and/or consultants to publish data potentially relating to our trade secrets, our agreements may contain certain limited publication rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. Because from time to time we expect to rely on third parties in the development, manufacture, and distribution of our products and provision of our services, we must, at times, share trade secrets with them. Despite employing the contractual and other security precautions described above, the need to share trade secrets increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. If any of these events occurs or if we otherwise lose protection for our trade secrets, the value of this information may be greatly reduced and our competitive position would be harmed. If we do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information may be jeopardized.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our trademarks or trade names, once registered, may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential partners or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Though these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names. Our efforts to enforce or protect our proprietary rights related to trademarks, trade names, trade secrets, domain names, copyrights, or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our financial condition or results of operations.
71
Moreover, any names we may propose to use with our product candidates in the U.S. must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA (or an equivalent administrative body in a foreign jurisdiction) objects to any of our proposed proprietary product names, it may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties, and be acceptable to the FDA. Similar requirements exist in Europe. Furthermore, in many countries, owning and maintaining a trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the owner of a senior trademark. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. If we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our future products.
Any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborations are subject to numerous risks, which may include that:
| · | collaborators have significant discretion in determining the efforts and resources that they will apply to collaborations; |
| · | collaborators may not pursue development and commercialization of our products or may elect not to continue or renew development or commercialization programs based on trial or test results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; |
| · | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates; |
| · | a collaborator with marketing, manufacturing, and distribution rights to one or more products may not commit sufficient resources to or otherwise not perform satisfactorily in carrying out these activities; |
| · | we could grant exclusive rights to our collaborators that would prevent us from collaborating with others; |
| · | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; |
| · | disputes may arise between us and a collaborator that causes the delay or termination of the research, development, or commercialization of our current or future products or that results in costly litigation or arbitration that diverts management attention and resources; |
72
| · | collaborations may be terminated, and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable current or future products; |
| · | collaborators may own or co-own intellectual property covering our products that results from our collaborating with them, and in such cases, we would not have the exclusive right to develop or commercialize such intellectual property; and |
| · | a collaborator’s sales and marketing activities or other operations may not be in compliance with applicable laws resulting in civil or criminal proceedings. |
Intellectual property discovered through government funded programs may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies. Compliance with such regulations may limit our exclusive rights and limit our ability to contract with non-U.S. manufacturers.
As of the date of this Registration Statement, neither our patents nor our product candidates are subject to march-in rights. However, some of our future patents may be generated through the use of U.S. government funding, and we may acquire or license in the future intellectual property rights that have been generated through the use of U.S. government funding or grants. Pursuant to the Bayh-Dole Act of 1980, the U.S. government has certain rights in inventions developed with government funding. These U.S. government rights include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right, under certain limited circumstances, to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (1) adequate steps have not been taken to commercialize the invention; (2) government action is necessary to meet public health or safety needs; or (3) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). If the U.S. government exercised its march-in rights in our future intellectual property rights that are generated through the use of U.S. government funding or grants, we could be forced to license or sublicense intellectual property developed by us or that we license on terms unfavorable to us, and there can be no assurance that we would receive compensation from the U.S. government for the exercise of such rights. The U.S. government also has the right to take title to these inventions if the grant recipient fails to disclose the invention to the government or fails to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. In addition, the U.S. government requires that any products embodying any of these inventions or produced through the use of any of these inventions be manufactured substantially in the U.S. This preference for U.S. industry may be waived by the federal agency that provided the funding if the owner or assignee of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the U.S. or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. industry may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property.
Risks Related to Legal and Regulatory Compliance Matters
Our relationships with customers, healthcare providers, including physicians, and third-party payors are subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Healthcare providers, including physicians, and third-party payors in the U.S. and elsewhere will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with healthcare professionals, principal investigators, consultants, customers and third- party payors subject us to various federal and state fraud and abuse laws and other healthcare laws, including, without limitation, the federal Anti-Kickback Statute, the federal civil and criminal false claims laws and the law commonly referred to as the Physician Payments Sunshine Act and regulations promulgated under such laws. These laws will impact, among other things, our clinical research, proposed sales, marketing and educational programs, and other interactions with healthcare professionals. In addition, we may be subject to patient privacy laws by both the federal government and the states in which we conduct or may conduct our business. The laws that will affect our operations include, but are not limited to:
73
| · | the federal Anti-Kickback Statute, which prohibits, among other things, individuals or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind in return for, or to induce, either the referral of an individual, or the purchase, lease, order or arrangement for or recommendation of the purchase, lease, order or arrangement for any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. A person does not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation; |
| · | the federal civil and criminal false claims laws, including, without limitation, the federal False Claims Act, which can be enforced by private citizens through civil whistleblower or qui tam actions, and civil monetary penalty laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from the federal government, including Medicare, Medicaid and other government payors, that are false or fraudulent or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim or to avoid, decrease or conceal an obligation to pay money to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. federal government. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of products for unapproved, and thus non-reimbursable, uses. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act; |
| · | HIPAA which created additional federal criminal statutes which prohibit, among other things, a person from knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| · | the federal transparency laws, including the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, medical devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program, with specific exceptions, to report annually to the CMMS, information related to: (i) payments or other “transfers of value’’ made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and (ii) ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations extended to include transfers of value made during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives; and |
| · | analogous state and foreign laws and regulations; state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures or drug pricing; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or that otherwise restrict payments that may be made to healthcare providers; and state and local laws that require the registration of pharmaceutical sales representatives. |
74
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participating in federal and state funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, contractual damages, diminished profits and future earnings, reputational harm and the curtailment or restructuring of our operations, any of which could harm our business.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. The shifting compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
Even if we obtain regulatory approval for PMN310, PMN442, PMN267 or any future product candidates, they will remain subject to ongoing regulatory oversight, which may result in significant additional expense.
Even if we obtain any regulatory approval for PMN310, PMN442 and PMN267 or any future product candidates, such product candidates will be subject to ongoing regulatory requirements applicable to research, development, testing, manufacturing, labeling, packaging, storage, advertising, promoting, sampling, record-keeping and submission of safety and other post-market information, among other things. Any regulatory approvals that we receive for PMN310, PMN442, PMN267 or any future product candidates may also be subject to Risk Evaluation and Mitigation Strategy, or REMS, limitations on the approved indicated uses for which the drug may be marketed or to the conditions of approval or requirements that we conduct potentially costly post-marketing testing and surveillance studies, including Phase 4 trials and surveillance to monitor the quality, safety and efficacy of the drug. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval. We will further be required to immediately report any serious and unexpected adverse events and certain quality or production problems with our products to regulatory authorities along with other periodic reports. Any new legislation addressing drug safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
In addition, drug manufacturers are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements and adherence to commitments made in the BLA or foreign marketing application. If we, or a regulatory authority, discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or if a regulatory authority disagrees with the promotion, marketing or labeling of that drug, a regulatory authority may impose restrictions relative to that drug, the manufacturing facility or us, including requesting a recall or requiring withdrawal of the drug from the market or suspension of manufacturing.
75
If we fail to comply with applicable regulatory requirements following approval of a product candidate, a regulatory authority may:
| · | issue a Form 483, an untitled letter or warning letter asserting that we are in violation of the law; |
| · | seek an injunction or impose administrative, civil or criminal penalties or monetary fines; |
| · | issue a safety alert, Dear Healthcare Provider letter, press release or other communication containing warnings or safety information about the product; |
| · | mandate corrections to promotional materials and labeling or issuance of corrective information; |
| · | suspend or withdraw regulatory approval; |
| · | suspend any ongoing clinical trials; |
| · | refuse to approve a pending marketing application or supplement to an approved application or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; |
| · | restrict the marketing or manufacturing of the drug; |
| · | seize or detain the drug or otherwise require the withdrawal of the drug from the market; |
| · | refuse to permit the import or export of products or product candidates; or |
| · | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize a product candidate, if approved, and harm our business, financial condition, results of operations and prospects.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions and civil or criminal penalties, private litigation or adverse publicity and could negatively affect our operating results and business.
We are subject to or affected by federal, state and foreign data protection laws and regulations which address privacy and data security. In the U.S., numerous federal and state laws and regulations, including HIPAA, as amended by HITECH, state data breach notification laws, state health information privacy laws and federal and state consumer protection laws, including Section 5 of the Federal Trade Commission Act, which govern the collection, use, disclosure and protection of health-related and other personal information, may apply to our operations and the operations of any future collaborators. In addition, we may obtain health information from third parties, including research institutions from which we obtain clinical trial data that are subject to privacy and security requirements under HIPAA, as amended by HITECH, and other privacy and data security laws. Depending on the facts and circumstances, we could be subject to significant administrative, civil and criminal penalties if we obtain, use or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. Further, various states have implemented similar privacy laws and regulations. For example, California also recently enacted the California Consumer Privacy Act of 2018, or CCPA. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used. The CCPA also provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA went into effect on January 1, 2020 and grants the California Attorney General the power to bring enforcement actions for violations beginning July 1, 2020. The CCPA has been amended from time to time, and it remains unclear what, if any, further modifications will be made to this legislation or how it will be interpreted. As currently written, the CCPA may impact our business activities and as a result may increase our compliance costs and potential liability. Many similar privacy laws have been proposed at the federal level and in other states.
76
Foreign data protection laws, including Regulation 2016/679, known as the General Data Protection Regulation, or GDPR, may also apply to health-related and other personal information data subjects in the EU or the United Kingdom, or UK. The GDPR went into effect on May 25, 2018. Companies that must comply with the GDPR face increased compliance obligations and risk, including robust regulatory enforcement of data protection requirements as well as potential fines for noncompliance of up to €20 million or 4% of annual global revenue of the noncompliance company, whichever is greater. The GDPR imposes numerous requirements for the collection, use, storage and disclosure of personal information of EU or UK data subjects, including requirements relating to providing notice to and obtaining consent from data subjects, personal data breach notification, cross-border transfers of personal information, and honoring and providing for the rights of EU or UK individuals in relation to their personal information, including the right to access, correct and delete their data.
Compliance with U.S. and foreign data protection laws and regulations could require us to take on more onerous obligations in our contracts, require us to engage in costly compliance exercises, restrict our ability to collect, use and disclose data, or in some cases, impact our or our partners’ or suppliers’ ability to operate in certain jurisdictions. Failure to comply with U.S. and foreign data protection laws and regulations could result in government investigations and/or enforcement actions, fines, civil or criminal penalties, private litigation or adverse publicity and could negatively affect our operating results and business.
Moreover, clinical trial subjects about whom we or any of our potential collaborators obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws or breached our contractual obligations, even if we are not found liable, could be expensive and time consuming to defend and could result in adverse publicity that could materially and adversely affect our business, financial condition, results of operations and prospects.
Even if we obtain FDA or EMA approval any of our product candidates in the U.S. or European Union, we may never obtain approval for or commercialize any of them in any other jurisdiction, which would limit our ability to realize their full market potential.
In order to market any products in any particular jurisdiction, we must establish and comply with numerous and varying regulatory requirements on a country-by-country basis regarding safety and efficacy.
Approval by the FDA in the U.S. or the EMA in the European Union does not ensure approval by regulatory authorities in other countries or jurisdictions. However, the failure to obtain approval in one jurisdiction may negatively impact our ability to obtain approval elsewhere. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not guarantee regulatory approval in any other country.
Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. In many jurisdictions outside the U.S., a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
77
Seeking foreign regulatory approval could result in difficulties and increased costs for us and require additional nonclinical studies or clinical trials, which could be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. We do not have any product candidates approved for sale in any jurisdiction, including in international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, our target market will be reduced and our ability to realize the full market potential of any product we develop will be unrealized.
Healthcare legislative or regulatory reform measures may have a negative impact on our business and results of operations.
The U.S. and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system. The U.S. government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
The Affordable Care Act substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum Medicaid rebate for both branded and generic drugs, expanded the 340B program, and revised the definition of AMP, which could increase the amount of Medicaid drug rebates manufacturers are required to pay to states. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service Medicaid utilization, to include the utilization of Medicaid managed care organizations as well and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the number of rebates due on those drugs. On February 1, 2016, CMMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016. Since that time, there have been significant ongoing efforts to modify or eliminate the Affordable Care Act.
The Affordable Care Act has been subject to challenges in the courts. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional and remanded the case to the Texas District Court to reconsider its earlier invalidation of the entire Affordable Care Act. An appeal was taken to the U.S. Supreme Court. On June 17, 2021, the U.S. Supreme Court ruled that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the U.S. Supreme Court did not rule on the constitutionality of the Affordable Care Act or any of its provisions.
Other legislative changes have been proposed and adopted since passage of the Affordable Care Act and we expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our product candidates, if approved, or additional pricing pressures.
78
Our business activities may be subject to the FCPA and similar anti-bribery and anti-corruption laws.
Our business activities may be subject to the FCPA, U.S. domestic bribery statutes, and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we may operate, including the U.K. Bribery Act of 2010. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, hospitals are owned and operated by the government, and doctors and other hospital employees would be considered foreign officials under the FCPA. There is no certainty that all of our employees, agents, contractors or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of our facilities, implementation of compliance programs and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our product candidates in one or more countries and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results and financial condition.
Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could significantly harm our business.
We are exposed to the risk of fraud or other misconduct by our employees, independent contractors, consultants, commercial partners and vendors. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with health care fraud and abuse laws and regulations in the U.S. and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our insurance policies are expensive and only protect us from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability, products liability and directors’ and officers’ insurance. We do not know, however, if we will be able to maintain insurance with adequate levels of coverage. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of PMN310 or any other product candidate. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
Risks Related to Our Business and Industry
We face significant competition in an environment of rapid technological and scientific change, and there is a possibility that our competitors may achieve regulatory approval before us or develop therapies that are safer or more effective than ours.
The development and commercialization of new drugs is highly competitive. Moreover, the AD field is characterized by strong competition and a strong emphasis on intellectual property. We may face competition with respect to any product candidates that we seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide.
79
Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
If approved, PMN310 will compete with therapies currently approved for the treatment of patients with AD, which have primarily been developed to treat the symptoms of AD rather than the underlying cause of the disease, such as memantine and cholinesterase inhibitors. PMN310 may also compete with one or more potentially disease-modifying therapeutics that target Aβ or amyloid plaques. Biogen’s aducanumab was approved by the FDA in June 2021 under the accelerated approval pathway, which allows for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint. Regulatory approval of aducanumab is pending in Europe and Japan. Other companies known to be developing therapies with Aβ/amyloid plaque-related targets include Alzheon, Inc., Alzinova AB, Chugai Pharmaceutical Co. Ltd., Cognition Therapeutics, Inc., Eisai Co., Ltd., Eli Lilly and Company, Grifols, S.A., KalGene Pharmaceuticals, Inc., Neurimmune AG, Novartis AG, Acumen Pharmaceuticals Inc., Prothena Biosciences, Inc., Roche Holding AG (including Genentech, its wholly owned subsidiary) and Wren Therapeutics, Inc. Additionally, PMN310, if approved, may also compete with other potential therapies intended to address underlying causes of AD that are being developed by several companies, including AbbVie Inc., AC Immune SA, Alector, Inc., Anavex Life Sciences Corp., Annovis Bio, Inc., Athira Pharma, Inc., Biohaven Pharmaceuticals, Inc., Cassava Sciences, Inc., Cortexyme, Inc., Denali Therapeutics, Inc., Johnson & Johnson (including Janssen, its wholly-owned subsidiary) and Takeda Pharmaceutical Co. Ltd.
Many of our current or potential competitors, either alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, nonclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved product candidates than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize product candidates that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any product candidates that we may develop. Furthermore, currently approved product candidates could be discovered to have application for treatment of AD, which could give such product candidates significant regulatory and market timing advantages over any of our product candidates. Our competitors also may obtain FDA, EMA or other regulatory approval for their product candidates more rapidly than we may obtain approval for ours from the FDA, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, product candidates or technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing any product candidates we may develop against competitors.
If our competitors market product candidates that are more effective, safer or less expensive than our product candidates, if approved, or that reach the market sooner than our product candidates, we may not achieve commercial success. In addition, the pharmaceutical industry is characterized by rapid technological change. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or product candidates developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
80
Risks Related to Ownership of Our Common Shares and Our Status as a U.S. Public Company
Investment in the Company’s Common Shares is speculative, involves risk, and there is no guarantee of a return.
There is no guarantee that the Common Shares will earn any positive return in the short term or long term. A holding of Common Shares is speculative and involves a high degree of risk and should be undertaken only by holders whose financial resources are sufficient to enable them to assume such risks and who have no need for immediate liquidity in their investment. A holding of Common Shares is appropriate only for holders who have the capacity to absorb a loss of some or all of their holdings.
If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our share price and trading volume could decline.
The trading market for our Common Shares will be influenced by the research and reports that equity research analysts publish about us and our business. As a newly public company, we anticipate having only limited research coverage by equity research analysts. Equity research analysts may elect not to provide research coverage of our Common Shares, and such lack of research coverage may adversely affect the market price of our Common Shares. In the event we do have equity research analyst coverage, we will not have any control over the analysts or the content and opinions included in their reports. The price of our shares could decline if one or more equity research analysts downgrade our shares or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our shares could decrease, which in turn could cause our shares price or trading volume to decline.
Concentration of ownership of our Common Shares among our existing executive officers, directors and principal shareholders may prevent new investors from influencing significant corporate decisions.
Based on their shareholdings as of March 25, 2022, our directors, executive officers and beneficial owners of greater than 5% of our outstanding shares and their respective affiliates will beneficially own, in the aggregate, approximately 27.59% of our outstanding Common Shares. As a result, these persons, acting together, would be able to significantly influence all matters requiring shareholder approval, including the election and removal of directors, any merger, consolidation, sale of all or substantially all of our assets or other significant corporate transactions.
Some of these persons or entities may have interests different than yours. For example, because many of these shareholders purchased their shares at prices substantially below the estimated public offering price and have held their shares for a longer period, they may be more interested in selling our Company to an acquirer rather than other investors, or they may want us to pursue strategies that deviate from the interests of other shareholders.
Our constating documents permit us to issue an unlimited amount of additional Common Shares or Preferred Shares, which may prevent a third-party takeover or cause our shareholders to experience dilution in the future.
Our constating documents authorize us to issue an unlimited number of Common Shares and an unlimited number of Preferred Shares. Our Board has the authority to cause us to issue additional Common Shares and Preferred Shares and to determine the special rights and restrictions of the shares of one or more series of our Preferred Shares, each without consent of our shareholders. The issuance of any such securities may result in a reduction of the book value or market price of our Common Shares. Given the fact that we have not achieved profitability or generated positive cash flow historically, and we operate in a capital-intensive industry with significant working capital requirements, we may be required to issue additional Common Shares or other securities that are dilutive to existing shareholders in the future in order to continue our operations. Our efforts to fund our intended business plan may result in dilution to existing shareholders. Further, any such issuances could result in a change of control or a reduction in the market price for our Common Shares. Additionally, the rights of the holders of Common Shares will be subject to, and may be adversely affected by, the rights of holders of any Preferred Shares that may be issued in the future. For example, Preferred Shares typically rank senior to Common Shares as to dividend rights, liquidation preference or both and may be convertible into Common Shares. Lastly, our ability to issue Preferred Shares could make it more difficult for a third-party to acquire a majority of our outstanding voting shares, particularly in the event we issue Preferred Shares with special voting rights, the effect of which may be to deprive our shareholders of a control premium that might otherwise be realized in connection with an acquisition of us.
Anti-takeover provisions in our governing documents and under Canadian Law could prevent or delay transactions that shareholders may favor.
Provisions of our governing documents and the CBCA may discourage, delay or prevent a merger or acquisition that shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for their Common Shares, and may also frustrate or prevent any attempt by shareholders to change the direction or management. For example, these provisions:
| · | require a 66 2/3% majority of shareholder votes cast in favor of a resolution to effect various amendments to the Articles of Incorporation of the Company, as amended (the “articles”); |
| · | require that in the event of shareholders of the Company vote via written resolution, that such resolution must be signed by all shareholders of the Company entitled to vote on that resolution; |
| · | establish advance notice requirements for nominations for election to the Board at any annual or special meeting of shareholders of the Company; |
| · | Any transaction in which a third party seeks to acquire our voting securities or equity securities that would result in the acquiror holding greater than 20% of the securities of that class may be governed by Multilateral Instrument 62-104—Take-Over Bids and Issuer Bids (the "Takeover Bid Rules") promulgated by the Canadian Securities Administrators. The "General Principles" of the Takeover Bid Rules and certain important aspects of the Takeover Bid Rules are described more fully in the section entitled "Description of the Registrant’s Securities to be Registered" on page 118; and |
81
| · | the Company is party to a shareholder rights plan agreement (the “Shareholder Rights Plan”) dated as of January 22, 2016 and amended and extended as of May 15, 2019 between the Company and Computershare Trust Company of Canada as rights agent to ensure, to the extent possible, that all shareholders of the Company are treated fairly in connection with any takeover bid for the Company. The Board authorized the issuance of one right in respect of each Common Share outstanding at 5:00 p.m. (ET) on January 22, 2016 and each Common Share issued thereafter. The rights will become exercisable if a person, together with their affiliates, associates and joint actors, acquires or announces an intention to acquire beneficial ownership of Common Shares which, when aggregated with its holdings, total 20% or more of our outstanding Common Shares (determined in the manner set out in the Shareholder Rights Plan). Following the acquisition of 20% or more of the outstanding Common Shares, each right held by a person other than the acquiring person and its affiliates, associates and joint actors would, upon exercise, entitle the holder to purchase that number of Common Shares at a substantial discount to the market price of the Common Shares at that time. Shareholder rights plans generally deter potential acquirors from making a take-over bid for securities of a company as it increases the cost of the securities of the company the acquiror is attempting to acquire. |
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation and these differences may make our Common Shares less attractive to investors.
We are incorporated under the federal laws of Canada, and, therefore, certain of the rights of holders of our shares are governed by Canadian law, including the provisions of the CBCA, and by our articles. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations and these differences may make our Common Shares less attractive to investors. The principal differences are described in the section entitled "Description of the Registrant’s Securities to be Registered" on page 118.
If we fail to attract and retain senior management and key scientific personnel, our business may be materially and adversely affected.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management and clinical and scientific personnel. We are highly dependent upon members of our senior management, particularly our CEO, Eugene Williams, as well as our senior scientists and other members of our management team. The loss of services of any of these individuals could delay or prevent the successful development of our product pipeline, initiation or completion of our planned clinical trials or the commercialization of our product candidates or any future product candidates.
Competition for qualified personnel in the biopharmaceutical field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our clinical development and if we initiate commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our current or future product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and breach of warranty. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
82
| · | decreased demand for our current or future product candidates; |
| · | injury to our reputation; |
| · | withdrawal of clinical trial participants; |
| · | costs to defend the related litigation; |
| · | diversion of management’s time and our resources; |
| · | substantial monetary awards to trial participants or patients; |
| · | regulatory investigations, product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | loss of revenue; and |
| · | the inability to commercialize our current or any future product candidates. |
If we are unable to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims, the commercialization of our current or any future product candidates we develop could be inhibited or prevented. We currently carry product liability insurance covering our clinical trials. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions and deductibles, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient funds to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If and when we obtain approval for marketing any of our product candidates, we intend to expand our insurance coverage to include the sale of such product candidate; however, we may be unable to obtain this liability insurance on commercially reasonable terms or at all.
We may explore strategic collaborations that may never materialize or may fail.
We may attempt to broaden the global reach of our platform by selectively collaborating with leading therapeutic companies and other organizations. As a result, we may periodically explore a variety of possible additional strategic collaborations in an effort to gain access to additional product candidates or resources. At the current time, we cannot predict what form such a strategic collaboration might take. In the event we do form such collaborations, we intend to retain significant economic and commercial rights to our programs in key geographic areas that are core to our long-term strategy. We are likely to face significant competition in seeking appropriate strategic collaborators, and strategic collaborations can be complicated and time consuming to negotiate and document. We may not be able to negotiate strategic collaborations on acceptable terms, or at all. We are unable to predict when, if ever, we will enter into any additional strategic collaborations because of the numerous risks and uncertainties associated with establishing them.
83
We are an “emerging growth company” and a “smaller reporting company” and, as a result of the reduced disclosure and governance requirements applicable to emerging growth companies and smaller reporting companies, our Common Shares may be less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act and we intend to take advantage of some of the exemptions from reporting requirements that are applicable to other public companies that are not emerging growth companies, including:
| · | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| · | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| · | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
| · | not being required to hold a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. |
We cannot predict if investors will find our Common Shares less attractive because we will rely on these exemptions. If some investors find our Common Shares less attractive as a result, there may be a less active trading market for our Common Shares and our share price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earlier of the last day of the fiscal year (i) following the fifth anniversary of the closing of our initial public offering, (ii) in which we have total annual gross revenue of at least $1.07 billion, or (iii) in which we are deemed to be a large accelerated filer, which means the market value of our Common Shares that are held by non-affiliates exceeds $700 million as of the prior June 30th, and the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to take advantage of the extended transition period to comply with new or revised accounting standards and to adopt certain of the reduced disclosure requirements available to emerging growth companies. As a result of the accounting standards election, we will not be subject to the same implementation timing for new or revised accounting standards as other public companies that are not emerging growth companies, which may make comparison of our financials to those of other public companies more difficult. As a result of these elections, the information that we provide in this Registration Statement may be different than the information you may receive from other public companies in which you hold equity interests. In addition, it is possible that some investors will find our Common Shares less attractive as a result of these elections, which may result in a less active trading market for our Common Shares and higher volatility in our share price.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies until the fiscal year following the determination that our voting and non-voting Common Shares held by non-affiliates is more than $250 million measured on the last business day of our second fiscal quarter, or our annual revenues are more than $100 million during the most recently completed fiscal year and our voting and non-voting Common Shares held by non-affiliates is more than $700 million measured on the last business day of our second fiscal quarter.
We have never paid dividends on our capital shares and we do not intend to pay dividends for the foreseeable future. Consequently, any gains from an investment in our Common Shares will likely depend on whether the price of our Common Shares increases.
We have never declared or paid any dividends on our Common Shares and do not intend to pay any dividends in the foreseeable future. We anticipate that we will retain all of our future earnings for use in the operation of our business and for general corporate purposes. Any determination to pay dividends in the future will be at the discretion of our Board. Accordingly, investors must rely on sales of their Common Shares after price appreciation, which may never occur, as the only way to realize any future gains on their investments.
84
We are subject to the continued listing criteria of the TSX and our failure to satisfy these criteria may result in a delisting of our Common Shares.
Our Common Shares are currently listed on the TSX. In order to maintain our listing on TSX, we must maintain certain financial and share distribution targets, including maintaining a minimum number of public shareholders. In addition to objective standards, the TSX may delist the securities of any issuer if, in its opinion, an issuer’s financial condition and/or operating results appear unsatisfactory, if it appears that the extent of public distribution or the aggregate market value of a security has become so reduced as to make continued listing on the TSX inadvisable, if the issuer sells or disposes of principal operating assets or ceases to be an operating company, if an issuer fails to comply with the listing requirements of TSX, or if any other event occurs or any condition exists which makes continued listing on the TSX, in the opinion of the TSX, inadvisable.
If the TSX delists our Common Shares, investors may face material adverse consequences, including, but not limited to, a lack of trading market for the Common Shares, reduced liquidity, decreased analyst coverage of the Company, and an inability for us to obtain additional financing to fund our operations.
We cannot assure you that our Common Shares will become listed on Nasdaq and, if listed, our failure to meet Nasdaq’s continued listing requirements could result in a delisting of our Common Shares.
Although we have submitted an application to list our Common Shares on Nasdaq, we cannot assure you that we will be able to meet the initial listing standards, including the minimum per share price and minimum capitalization requirements, or that we will be able to maintain a listing of our Common Shares on any such trading venue. If, after listing, we fail to satisfy Nasdaq’s continued listing requirements, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our Common Shares. Such a delisting would likely have a negative effect on the price of our Common Shares and would impair your ability to sell or purchase our Common Shares when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our Common Shares to become listed again, stabilize the market price or improve the liquidity of our Common Shares, prevent our Common Shares from dropping below the required minimum bid price or prevent future non-compliance with Nasdaq listing requirements.
Our internal controls over financial reporting may not be effective, which could have a material and adverse effect on our business.
The Company is subject to reporting and other obligations under applicable Canadian securities laws and rules of any stock exchange on which the Common Shares are listed, including NI 52-109, and upon effectiveness of this Registration Statement, will be subject to U.S. securities reporting and regulatory requirements. These reporting and other obligations place significant demands on our management, administrative, operational and accounting resources. If we are unable to accomplish any such necessary objectives in a timely and effective manner, our ability to comply with our financial reporting obligations and other rules applicable to reporting issuers could be impaired. Moreover, any failure to maintain effective internal controls could cause us to fail to satisfy our reporting obligations or result in material misstatements in our financial statements. If we cannot provide reliable financial reports or prevent fraud, our reputation and operating results could be materially adversely affected, which could also cause investors to lose confidence in our reported financial information, which could result in a reduction in the trading price of the Common Shares.
The Company does not expect that its disclosure controls and procedures and internal controls over financial reporting will prevent all error or fraud. A control system, no matter how well-designed and implemented, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Due to the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues within an organization are detected. The inherent limitations include the realities that judgments in decision making can be faulty, and that breakdowns can occur because of simple errors or mistakes. Controls can also be circumvented by individual acts of certain persons, by collusion of two or more people or by management override of the controls. Due to the inherent limitations in a control system, misstatements due to error or fraud may occur and may not be detected in a timely manner or at all.
85
The elimination of monetary liability against our directors, officers, and employees under Canadian law and the existence of indemnification rights for our obligations to our directors, officers, and employees may result in substantial expenditures by us and may discourage lawsuits against our directors, officers, and employees.
Our by-laws provide that, subject to the CBCA, we may indemnify a director or officer or a former director or officer or a corporation of which the we are or were a shareholder or creditor and their heirs and legal representatives of such person against all costs, charges, and expenses including and amount to be paid to settle an action or satisfy a judgment, reasonably incurred in respect of any civil, criminal or administrative action or proceeding to which they are made a party by reason of being or having been a director or officer of us or a director or officer of any such corporation. Each director and officer upon being elected and appointed shall be deemed to have contracted with us on the terms of this indemnity. The failure of a director or officer to comply with the provisions of the CBCA or the articles or the by-laws shall not invalidate any indemnity to which they are entitled under the by-laws.
We may also have contractual indemnification obligations under any future employment agreements with our officers or agreements entered into with our directors. The foregoing indemnification obligations could result in us incurring substantial expenditures to cover the cost of settlement or damage awards against directors and officers, which we may be unable to recoup. These provisions and the resulting costs may also discourage us from bringing a lawsuit against directors and officers for breaches of their fiduciary duties, and may similarly discourage the filing of derivative litigation by our shareholders against our directors and officers even though such actions, if successful, might otherwise benefit us and our shareholders.
There may be difficulty in enforcing judgments and effecting service of process on directors and officers that are not citizens of the U.S.
We are incorporated under the CBCA and some of our directors and officers reside outside of the U.S., in Canada. Consequently, it may not be possible for an investor to effect service of process within the U.S. on us or those persons. Furthermore, it may not be possible for an investor to enforce judgments obtained in U.S. courts based upon the civil liability provisions of U.S. federal securities laws or other laws of the U.S. against us or those persons. There is doubt as to the enforceability, in original actions in Canadian courts, of liabilities based upon U.S. federal securities laws and as to the enforceability in Canadian courts of judgments of U.S. courts obtained in actions based upon the civil liability provisions of the U.S. federal securities laws. Therefore, it may not be possible to enforce those actions against us and certain of our directors and officers.
If we are characterized as a passive foreign investment company (“PFIC”), U.S. Holders may be subject to adverse U.S. federal income tax consequences.
Based on our current operations, income, assets and certain estimates and projections, including as to the relative values of our assets, including goodwill, which is based on the expected price of our Common Shares, we were not a PFIC for the 2021 taxable year and do not expect to be a PFIC for the 2022 taxable year.
However, we must make an annual determination as to whether we are a PFIC based on the types of income we earn and the types and value of our assets from time to time, all of which are subject to change. Therefore, we cannot assure you that we will not be a PFIC for our current taxable year or any future taxable year. A non-U.S. corporation generally will be considered a PFIC for any taxable year if either (1) at least 75% of its gross income is passive income or (2) at least 50% of the value of its assets (based on an average of the quarterly values of the assets during a taxable year) is attributable to assets that produce or are held for the production of passive income. The market value of our assets may be determined in large part by the market price of the Common Shares, which is likely to fluctuate. In addition, the composition of our income and assets will be affected by how, and how quickly, we use any cash that we raise. If we were to be treated as a PFIC for any taxable year during which you hold Common Shares, certain adverse U.S. federal income tax consequences could apply to U.S. Holders.
For purposes of this discussion, a “U.S. Holder” is a holder who, for U.S. federal income tax purposes, is a beneficial owner of Common Shares, and who is: (i) an individual who is a citizen or individual resident of U.S.; (ii) a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the U.S., any state therein or the District of Columbia; (iii) an estate the income of which is subject to U.S. federal income taxation regardless of its source; or (iv) a trust if (1) a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (2) the trust has a valid election in effect to be treated as a U.S. person under applicable U.S. Treasury Regulations.
86
General Risk Factors
We will incur increased costs and demands upon management as a result of being a public company.
As a public company listed in the U.S., we will incur significant additional legal, accounting and other costs. These additional costs could negatively affect our financial results. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including regulations implemented by the SEC and Nasdaq, may increase legal and financial compliance costs and make some activities more time-consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies.
We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If notwithstanding our efforts to comply with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
Failure to comply with these rules might also make it more difficult for us to obtain some types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our Board, on committees of our Board or as members of senior management.
Comprehensive tax reform legislation could adversely affect our business and financial condition.
The rules dealing with U.S. federal, state and local income taxation are constantly under review by persons involved in the legislative process and by the Internal Revenue Service and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect us or holders of our Common Shares.
Additional changes to U.S. federal income tax law are currently being contemplated, and future changes in tax laws could have a material adverse effect on our business, cash flow, financial condition or results of operations. It cannot be predicted whether, when, in what form, or with what effective dates, new tax laws may be enacted, or regulations and rulings may be enacted, promulgated or issued under existing or new tax laws, which could result in an increase in our or our stockholders’ tax liability or require changes in the manner in which we operate in order to minimize or mitigate any adverse effects of changes in tax law or in the interpretation thereof.
87
Our business and operations would suffer in the event of computer system failures, cyberattacks or a deficiency in our cybersecurity or a natural disaster.
Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, natural disasters, terrorism, war, telecommunication and electrical failures, cyberattacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyberattacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material legal claims and liability and damage to our reputation, and the further development of our product candidates could be delayed.
Disruptions at the FDA, the SEC and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs or biologics to be reviewed and approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including most recently from December 22, 2018 to January 25, 2019, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
FDA and regulatory authorities outside the U.S. may adopt policy measures in response to the COVID-19 pandemic and may experience delays in their regulatory activities. In response to the COVID-19 pandemic, on March 10, 2020 the FDA announced its intention to postpone most inspections of foreign manufacturing facilities and products while local, national and international conditions warrant. On March 18, 2020, the FDA announced its intention to temporarily postpone routine surveillance inspections of domestic manufacturing facilities and provided guidance regarding the conduct of clinical trials, which the FDA continues to update. As of June 23, 2020, the FDA noted it was continuing to ensure timely reviews of applications for medical products during the COVID-19 pandemic in line with its user fee performance goals and conducting mission critical domestic and foreign inspections to ensure compliance of manufacturing facilities with FDA quality standards. As of July 2020, utilizing a rating system to assist in determining when and where it is safest to conduct such inspections based on data about the virus’ trajectory in a given state and locality and the rules and guidelines that are put in place by state and local governments, FDA is either continuing to, on a case-by-case basis, conduct only mission critical inspections, or, where possible to do so safely, resuming prioritized domestic inspections, which generally include pre-approval inspections. Foreign pre-approval inspections that are not deemed mission-critical remain postponed, while those deemed mission-critical will be considered for inspection on a case-by-case basis. FDA will use similar data to inform resumption of prioritized operations abroad as it becomes feasible and advisable to do so. The FDA may not be able to maintain this pace and delays or setbacks are possible in the future. Should FDA determine that an inspection is necessary for approval, and an inspection cannot be completed during the review cycle due to restrictions on travel, FDA has stated that it generally intends to issue a complete response letter. Further, if there is inadequate information to make a determination on the acceptability of a facility, FDA may defer action on the application until an inspection can be completed. Additionally, regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic and may experience delays in their regulatory activities.
88
If a prolonged government shutdown occurs, or if global health concerns prevent the FDA or other regulatory authorities from conducting business as usual or conducting inspections, reviews or other regulatory activities, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. Portions of our future clinical trials may be conducted outside of the U.S. and unfavorable economic conditions resulting in the weakening of the U.S. dollar would make those clinical trials more costly to operate. Furthermore, a severe or prolonged economic downturn, including a recession or depression resulting from the current COVID-19 pandemic or other public health crises, weather catastrophe, acts of terrorism, war (such as the military conflict between Russia and Ukraine), threats of terrorist attacks or war, political disruption or other events outside of our control could result in a variety of risks to our business, including, among other things, weakened demand for our product candidates or any future product candidates, if approved, and our ability to raise additional capital when needed on acceptable terms, if at all. Any of the foregoing could seriously harm our business, and we cannot anticipate all of the ways in which the political or economic climate and financial market conditions could seriously harm our business.
89
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
All references in this management’s discussion and analysis of financial condition and results of operations, or MD&A, to the “Company”, “ProMIS”, “we”, “us”, or “our” refer to ProMIS Neurosciences Inc., unless otherwise indicated or the context requires otherwise. The following MD&A is prepared as of April 4, 2022 for the year ended December 31, 2021 and 2020 and should be read in conjunction with the audited consolidated financial statements for the year ended December 31, 2021 and 2020 (the “Financial Statements”), which have been prepared by management in accordance with United States generally accepted accounting principles (“U.S. GAAP”) as issued by the Financial Accounting Standards Board (“FASB”). All dollar amounts refer to United States dollars, except as stated otherwise.
Overview
We are applying our patented technology platform to build a portfolio of antibody therapies, therapeutic vaccines, and other antibody-based therapies in neurodegenerative diseases and other mis-folded diseases, including AD, MSA, and ALS. The Company also plans to investigate additional synucleinopathies, including PD, DLB, FTLD, PSP, CBD and schizophrenia. These diseases share a common biologic cause – misfolded versions of proteins, that otherwise perform a normal function, become toxic and kill neurons, resulting in disease. ProMIS’ technology platform is an example of the advances in drug discovery enabled by computational power, in silico discovery, and/or artificial intelligence. We believe this platform provides a potential advantage in selectively targeting the toxic misfolded proteins with therapeutics or detecting them with diagnostics.
We are developing a pipeline of antibodies aimed at selectively targeting misfolded toxic forms of proteins that drive neurodegenerative diseases without interfering with the essential functions of the same properly folded proteins. Our product candidates are PMN310, PMN442, and PMN267. Our lead product candidate is PMN310, a monoclonal antibody designed to treat AD by selectively targeting the toxic misfolded form of Aβ. In light of research suggesting that misfolded toxic a-syn is a primary driver of disease, our second lead product candidate, PMN442, shows robust binding to a-syn oligomers and seeding fibrils in preclinical studies, with negligible binding to a-syn monomers and physiologic tetramers which are required for normal neuronal function. PMN267 is our third lead product candidate, which has been shown in preclinical studies to selectively recognize misfolded, cytoplasmic TDP-43 aggregates without interacting with endogenous normal TDP-43. TDP-43 is believed to play an important role in the development of ALS. We also have a number of development programs as discussed in the Business section of this Registration Statement.
We are incorporated under the CBCA and located at 1920 Yonge Street, Toronto, Ontario. We are traded on the TSX under the symbol PMN and on the OTCQB Venture Market under the symbol ARFXF. We have a wholly-owned U.S. subsidiary, ProMIS USA, which was incorporated in January 2016 in the State of Delaware. ProMIS USA has had no activity and has no financial impact on our Financial Statements. Since our inception, we have devoted substantially all of our resources to developing our platform technologies, building our intellectual property portfolio, business planning, raising capital and providing general and administrative support for these operations. We have principally financed our operations through private placements of common shares and warrants and convertible debt.
We have incurred significant operating losses since inception. Our ability to generate product revenue sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of our product candidates and any future product candidates. Our net losses were $9.8 million and $4.3 million for the years ended December 31, 2021 and December 31, 2020, respectively. As of December 31, 2021, we had an accumulated deficit of $62.2 million. We expect to continue to incur net losses for the foreseeable future and expect our research and development expenses, general and administrative expenses and capital expenditures to increase. In particular, we expect our expenses to increase as we continue our development of, and seek regulatory approvals for, our product candidates, as well as initiate clinical trials, hire additional personnel, pay fees to outside consultants, lawyers and accountants, and incur other increased costs associated with being a public company. In addition, if we obtain marketing approval for any product candidates, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. We may also incur expenses in connection with the in-licensing or acquisition of additional product candidates. Furthermore, upon the effectiveness of this Registration Statement, we expect to incur additional costs associated with operating as a public company in the United States, including significant legal, accounting, investor relations, compliance and other expenses that we did not incur as a public Canadian company.
As a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through the sale of equity, debt financings, or other capital sources, which may include collaborations with other companies or other strategic transactions. We may be unable to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as and when needed, we may have to significantly delay, reduce or eliminate the development and commercialization of one or more of our product candidates or delay our pursuit of potential in-licenses or acquisitions.
90
Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
We expect that our cash of $16.9 million as of December 31, 2021, will fund our operating expenses and capital expenditure requirements through the end of 2023. We have based this estimate on assumptions that may prove to be wrong, and we could exhaust our available capital resources sooner than we expect. See “Liquidity and Capital Resources.”
Components of Operating Results
Revenue
We have not generated any revenue since our inception and do not expect to generate any revenue from the sale of our products in the near future, if at all. If our product candidates are successful and result in marketing approval or if we enter into collaboration or license agreements with third parties, we may generate revenue in the future from a combination of product sales or payments from such collaboration or license agreements.
Operating Expenses
Research and Development Expenses
Research and development expenses consist primarily of costs incurred in connection with the development and research of our platform technologies, as well as unrelated discovery program expenses. We expense research and development costs in the periods in which they are incurred. These expenses include:
| · | employee-related expenses, including salaries, related benefits and share-based compensation expense, for employees engaged in research and development activities; |
| · | external research and development expenses incurred under arrangements with third parties, such as contract research organizations or CROs, and consultants; |
| · | the cost of acquiring, developing, and manufacturing clinical study materials; and |
| · | costs associated with preclinical and clinical activities and regulatory operations. |
We enter into consulting, research, and other agreements with commercial entities, researchers, universities, and others for the provision of goods and services. Such arrangements are generally cancelable upon reasonable notice and payment of costs incurred. Costs are considered incurred based on an evaluation of the progress to completion of specific tasks under each contract using information and data provided by the respective vendors, including our clinical sites. These costs consist of direct and indirect costs associated with our platform technologies, as well as fees paid to various entities that perform certain research on our behalf. Depending upon the timing of payments to the service providers, we recognize prepaid expenses or accrued expenses related to these costs. These accrued or prepaid expenses are based on management’s estimates of the work performed under service agreements, milestones achieved, and experience with similar contracts. We monitor each of these factors and adjust estimates accordingly. See “Item 1A. Risk Factors” in this document.
Research and development activities account for a significant portion of our operating expenses. We expect our research and development expenses to increase substantially for the foreseeable future as we continue to implement our business strategy, which includes advancing our platform technologies through clinical development as well as other product candidates into clinical development, expanding our research and development efforts, including hiring additional personnel to support our research efforts, our clinical and product development efforts, and seeking regulatory approvals for our product candidates that successfully complete clinical trials.
91
We use our personnel and infrastructure resources across multiple research and development programs directed toward identifying and developing product candidates. Our direct research and development expenses consist primarily of external costs, including fees paid to consultants, contractors and CROs in connection with our development activities and the cost of acquiring, developing, and manufacturing clinical study materials.
General and Administrative Expenses
General and administrative expenses consist primarily of personnel costs including salary, bonus, employee-benefits and share-based compensation, costs incurred in development and protection of intellectual property, professional service fees, and other general overhead and facility costs, (including rent) depreciation and amortization. We expect our general and administrative expenses to increase substantially for the foreseeable future as we increase our administrative function to support the growth of the business and its continued research and development activities.
Other (Expense) Income
Other (expense) income consists primarily of interest expense on our Debentures and changes in the fair value of our financial instruments.
Result of Operations
Years Ended December 31, 2021 and 2020
The following table summarizes our results of operations for the periods presented:
Years Ended December 31, | ||||||||||||
| 2021 | 2020 | Change | ||||||||||
| Operating expenses | ||||||||||||
| Research and development | $ | 4,627,386 | $ | 2,224,650 | $ | 2,402,736 | ||||||
| General and administrative | 3,663,707 | 2,026,957 | 1,636,750 | |||||||||
| Total operating expenses | 8,291,093 | 4,251,607 | 4,039,486 | |||||||||
| Loss from operations | (8,291,093 | ) | (4,251,607 | ) | 4,039,486 | |||||||
| Other (expense)/income | (1,499,013 | ) | 1,327 | 1,500,340 | ||||||||
| Net loss | $ | (9,790,106 | ) | $ | (4,250,280 | ) | $ | 5,539,826 | ||||
Research and Development Expenses
The following table summarizes the period-over-period changes in research and development expenses for the periods presented:
| Years Ended December 31, | ||||||||||||
| 2021 | 2020 | Change | ||||||||||
| Direct research and development expenses by program | ||||||||||||
| PMN310 | $ | 2,654,430 | $ | 530,015 | $ | 2,124,415 | ||||||
| ALS | 376,656 | 19,193 | 357,463 | |||||||||
| Platform and other programs | 346,655 | 176,173 | 170,482 | |||||||||
| Indirect research and development expenses: | ||||||||||||
| Personnel related expense, including share-based compensation | 741,121 | 1,358,575 | (617,454 | ) | ||||||||
| Consulting expense | 462,699 | 129,013 | 333,686 | |||||||||
| Other operating costs | 45,825 | 11,681 | 34,144 | |||||||||
| Total research and development expenses | $ | 4,627,386 | $ | 2,224,650 | $ | 2,402,736 | ||||||
Research and development expenses increased by $2.4 million, or 108%, for the year ended December 31, 2021 compared to the year ended December 31, 2020. This increase is attributable to a $2.7 million increase in direct research and development expenses related to a $2.1 million increase in spending on our lead program, PMN310, largely attributable to $1.8 million of expenses on pre-clinical preparation costs and $0.3 million on external research costs, a $0.4 million expenses on external research costs on ALS portfolio projects and a $0.2 million increase in spending on our platform technology and other projects. The $0.3 million increase in consulting expense relates to various consultants advising on the preparation of the IND and design of preclinical and clinical trials. The increases were partially offset by a decrease of $0.6 million in personnel related expenses due to a reduction in management compensation and the attrition of contracted staff.
92
General and Administrative Expenses
The following table summarizes the period-over-period changes in general and administrative expenses for the periods presented:
| Years Ended December 31, | ||||||||||||
| 2021 | 2020 | Change | ||||||||||
| Personnel related, including share-based compensation | $ | 966,125 | $ | 736,529 | $ | 229,596 | ||||||
| Professional and consulting fees | 2,203,685 | 973,979 | 1,229,706 | |||||||||
| Patent expense | 438,935 | 256,126 | 182,809 | |||||||||
| Facility-related and other | 54,962 | 60,323 | (5,361 | ) | ||||||||
| Total general and administrative expenses | $ | 3,663,707 | $ | 2,026,957 | $ | 1,636,750 | ||||||
General and administrative expenses increased by $1.6 million, or 81%, for the year ended December 31, 2021 compared to the year ended December 31, 2020. The increase was primarily due to $0.6 million from the expensing of share issue costs associated with the August 2021 financing, one-time costs of $0.4 million related to a potential listing on a stock exchange in the United States, which such listing is not assured, increased consulting fees of $0.4 million and legal and patent fees of $0.3 million, and an increase in foreign exchange losses of $0.2 million, offset by a decrease of $0.3 million in investor relations expenses due to decreased investor relations activities.
Other Expense/Income
Other expense increased by $1.5 million for the year ended December 31, 2021 compared to the year ended December 31, 2020. The increase was primarily due to $0.4 million of interest expense incurred on the Debentures and $1.1 million due to the change in fair value of financial instruments.
Liquidity and Capital Resources
Sources of Liquidity
We are a development stage company as we have had minimal recurring revenues to date and do not expect to have significant revenues until we are able to sell a product candidate after obtaining applicable regulatory approvals or we establish collaborations that provide funding, such as licensing fees, milestone payments, royalties, research funding or otherwise. Operations have been financed since inception, through the sale of equity and debt securities and the conversion of common share purchase warrants and share options. Our objectives, when managing capital, are to ensure there are sufficient funds available to carry out our research, development and eventual commercialization programs. When we have excess funds, we manage our liquidity risk by investing in highly liquid corporate and government bonds with staggered maturities to provide regular cash flow for current operations. We do not hold any asset-backed commercial paper and our cash is not subject to any external restrictions. We also manage liquidity risk by frequently monitoring actual and projected cash flows. The Board reviews and approves the Company’s operating and capital budgets, as well as any material transactions not in the ordinary course of business. The majority of our accounts payable and accrued liabilities have maturities of less than three months. We are dependent on our ability to generate revenues from our products or secure additional financing in order to continue our research and development activities and meet our ongoing obligations.
93
In March 2020, we announced we had received approval from the TSX to amend the exercise price of an aggregate of 44,182,530 outstanding warrants. The exercise price of the warrants repriced was C$0.13 per share, effective April 8, 2020 and expiring on May 22, 2020. The warrants repriced ranged in exercise prices of C$0.285 to C$0.48 At the end of the warrant repricing period, all unexercised warrants reverted to the original exercise price. All other terms of the warrants remained unchanged. There was a total of 44,182,530 warrants repriced and of the repriced warrants, 9,532,276 were exercised during the repricing period ended May 22, 2020, for total proceeds of $0.9 million.
In November 2020, we closed on a special warrant financing ("Special Warrants"). We issued 16,219,581 Special Warrants for gross proceeds of $1.5 million ($1.3 million, net of issuance costs). Each Special Warrant is exercisable, without payment of any additional consideration by the holder, into one common share and one transferable common share warrant (“Warrant”). Each Warrant entitles the holder to acquire one common share at an exercise price at C$0.20 per warrant share for a period of 60 months until November 4, 2025. Each Special Warrant will automatically convert at the earlier of the date that is (i) the third business day after a receipt for a final prospectus qualifying the distribution of the shares and warrants issuable upon the conversion of the Special Warrants and (ii) four months and one day after the issue date of the Special Warrants. In March 2021, the Special Warrants automatically converted into 16,219,581 common shares and 16,219,581 common share warrants.
In March 2021, we completed a $7.0 million private placement the Debentures. The Debentures are convertible into common shares at the option of the holder at a conversion price of $0.10 and accrue interest at 1% per annum, which is payable annually. At the Company’s election, accrued interest may be paid in cash or common shares (such number of shares determined by dividing the interest due by the five-day VWAP of the common shares. The Debentures mature on March 22, 2026. Prior to the maturity date, the Company is entitled to convert of the Debentures at the conversion price upon raising an aggregate of $50 million in equity and/or debt. On the maturity date, the Company may redeem the outstanding principal amount of the Debentures in either cash or common shares (at the then five-day VWAP less a 10% discount) or a combination thereof.
In August 2021, we announced the closing of a public offering of 125,781,250 common share units at a price of $0.16 per common share unit for gross proceeds of $20.1 million. Each common share unit consisted of one common share and one-quarter common share purchase warrant. Each whole warrant entitles the holder thereof to purchase one common share at an exercise price of $0.21 per share at any time for five years. The warrants contain an acceleration clause allowing the Company to accelerate the expiry date of the warrants to 30 days following a time period during which the common share VWAP exceeds a TSX trading price of $0.63 for ten consecutive trading days.
We incurred a net loss of $9.8 million for the year ended December 31, 2021 and reported an accumulated deficit of $62.2 million. We expect available funds will be sufficient to fund our operating expenses through the end of 2023. However, additional funding will be necessary to fund future research, pre-clinical and clinical activities. We will seek additional funding through public financings, debt financings, collaboration agreements, strategic alliances and licensing agreements. Although we have been successful in raising capital in the past, there is no assurance of success in obtaining such additional financing on terms acceptable to us, if at all, and there is no assurance that we will be able to enter into collaborations or other arrangements. If we are unable to obtain funding, it could force us to delay, reduce or eliminate research and development programs and product portfolio expansion or commercialization efforts. These potential delays, reductions and eliminations could adversely affect future business prospects, and the ability to continue operations.
Cash Flows
The following table summarizes our sources and uses of cash for the periods presented:
| Years Ended December 31, | ||||||||||||
| 2021 | 2020 | Change | ||||||||||
| Net cash used in operating activities | $ | 9,305,383 | ) | $ | (3,232,532 | ) | $ | (6,072,851 | ) | |||
| Net cash provided by (used in) investing activities | 94,618 | (83,089 | ) | 177,707 | ||||||||
| Net cash provided by financing activities | 25,522,801 | 2,864,918 | 22,657,883 | |||||||||
| Effect of exchange rates on cash | (175,018 | ) | (24,235 | ) | (150,783 | ) | ||||||
| Net increase (decrease) in cash | $ | 16,137,018 | $ | (474,938 | ) | $ | 16,611,956 | |||||
Cash Flows from Operating Activities
Cash used in operating activities was $9.3 million for the year ended December 31, 2021, which consisted of a net loss of $9.8 million, partially offset by $2.0 million in non-cash charges and a net change of $1.6 million in our net operating assets and liabilities. The non-cash charges primarily consisted of the change in fair value of finanical instruments of $1.1 million, share-based compensation of $0.5 million and $0.4 million for amortization of debt discount. Changes in cash flows related to operating assets and liabilities primarily consisted of a $1.4 million decrease in deferred compensation for management.
94
Cash used in operating activities was $3.2 million for the year ended December 31, 2020, which consisted of a net loss of $4.3 million, partially offset by $0.4 million in non-cash charges and a net change of $0.7 million in our net operating assets and liabilities. The non-cash charges primarily consisted of share-based compensation of $0.4 million. Changes in cash flows related to operating assets and liabilities primarily consisted of a $0.6 million increase in deferred compensation for management.
Cash Flows from Investing Activities
Cash provided by investing activities was $0.1 million for the year ended December 31, 2021, which related primarily to the sale of property and equipment.
Cash used in investing activities was $0.1 million for the year ended December 31, 2020, which related primarily to purchases of property and equipment.
Cash Flows from Financing Activities
Cash provided by financing activities was $25.5 million for the year ended December 31, 2021, which consisted of $18.6 million of proceeds from the issuance of common share units and $6.9 million of proceeds from the issuance of the Debentures.
Cash provided by financing activities was $2.9 million for the year ended December 31, 2020, which consisted of $1.6 million of proceeds from the issuance of common shares related to the exercise of warrants and $1.3 million of proceeds from the issuance of the Special Warrants.
Critical Accounting Policies and Estimates
Our MD&A is based on our Financial Statements, which have been prepared in conformity with U.S GAAP. The preparation of these Financial Statements in conformity with U.S. GAAP requires management to make certain estimates, judgements and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the Financial Statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgement about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from those estimates under different assumptions or conditions.
We believe that the following critical accounting estimates are most important to understanding and evaluating our reported financial results.
Share-based Compensation
Share-based compensation expense related to share awards granted to employees, directors and non-employees is recognized based on the grant-date estimated fair values of the awards using the Black Scholes option pricing model ("Black Scholes"). The value is recognized as expense ratably over the requisite service period, which is generally the vesting term of the award. We adjust the expense for actual forfeitures as they occur. Share based compensation expense is classified in the accompanying consolidated statements of operations and comprehensive loss based on the function to which the related services are provided.
Black-Scholes requires a number of assumptions, of which the most significant are expected volatility, expected option term (the time from the grant date until the options are exercised or expire) and risk-free rate. Expected volatility is determined using the historical volatility for the Company. The risk-free interest rate is based on the yield of Canadian government bonds with a remaining term equal to the expected life of the option. Expected dividend yield is zero because we have never paid cash dividends on common shares, and we do not expect to pay any cash dividends in the foreseeable future.
Embedded Derivatives
In March 2021, the Company completed a $7.0 million private placement of Debentures (see Note 10 to our Financial Statements). The Debentures contained certain embedded features that were assessed for derivative accounting pursuant to ASC 815. Pursuant to ASC 815, we accounted for the conversion feature as derivative liability and recorded the embedded conversion feature at fair value and adjust the instrument to fair value at each reporting with the difference recorded in earnings. The conversion feature was initially measured and at each reporting period using a scenario-based valuation method using a Monte Carlo model. Significant estimates are required to determine expected volatility and risk-free interest rate. The Company determines these assumptions mainly by reference to historical experience.
95
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our Consolidated Financial Statements appearing at the end of this registration statement.
Contractual Arrangements and Commitments
In February 2009, we entered into an agreement with UBC to further the development of, and to commercialize certain technology developed in part by the Company’s Chief Scientific Officer. The agreement was amended and restated in October 2015. Under the amended and restated agreement, we are committed to make royalty payments based on revenue earned from the licensed technology. An annual license fee is payable over the term of the agreement. The agreement remains effective unless terminated under the provisions of the agreement.
In April 2006, and additional amendments through November 2013, we entered into an agreement with the UHN, Toronto, to license certain technology and related intellectual property. Under the agreement, we are committed to make milestone payments of up to C$0.6 million based on the successful outcomes of predefined clinical and regulatory outcomes, and royalty payments based on revenue earned from licensed technology.
We indemnify our directors and officers against any and all claims and losses reasonably incurred in the performance of their service to the Company. We have and maintain liability insurance for our directors and officers.
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
In the normal course of business, we are exposed to a number of financial risks that can affect our operating performance. These risks are credit risk, liquidity risk and market risk. Our overall risk management program and prudent business practices seek to minimize any potential adverse effects on the Company’s financial performance.
Credit Risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk consist primarily of cash and short-term investments. We manage our exposure to credit losses by placing our cash with accredited financial institutions, which at times, may exceed federally insured limits, and when we have excess funds, such funds are invested in high-quality government and corporate issuers with low credit risk. Cash held is not subject to any external restrictions. As of December 31, 2021, a hypothetical 10% relative change in interest rates would not have a material impact on our Financial Statements.
Liquidity Risk
Our exposure to liquidity risk is dependent on purchasing obligations and raising funds to meet commitments and sustain operations. We are a pre-revenue development stage company, and we rely on external fundraising to support our operations. We also manage liquidity risk by continuously monitoring actual and projected cash flows. Our Board reviews and approves the Company’s operating budget, as well as any material transaction.
Foreign Currency Exchange Risk
We are exposed to foreign exchange risk on our US dollar denominated cash and US dollar denominated liabilities. As of December 31, 2021, we held USD $16.9 million of cash and prepaid expenses and USD $11.4 million of accounts payable, accrued liabilities, convertible debt, derivative and warrant liability. A 10% change in the USD exchange rate on the December 31, 2021 balances would impact net loss by $0.6 million.
Inflation Risk
Inflation generally affects us by increasing our cost of labor, outside consultants and CRO’s. We do not believe that inflation had a material effect on our business, financial condition or results of operations during the years ended December 31, 2021 or 2020.
96
The Company does not own or lease any material properties.
ITEM 4. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth the expected beneficial ownership of the Company’s Common Shares as of March 25, 2022 for (i) each member of the Board, (ii) each named executive officer (as defined below), (iii) each person known to the Company to be the beneficial owner of more than 5% of the Company’s securities and (iv) the members of the Board and the executive officers of the Company as a group. Beneficial ownership is determined according to the rules of the SEC. Generally, a person has beneficial ownership of a security if the person possesses sole or shared voting or investment power of that security, including any securities of which a person has the right to acquire beneficial ownership within 60 days. Information with respect to beneficial owners of more than 5% of the Company’s securities is based on completed questionnaires and related information provided by such beneficial owners as of March 25, 2022. Except as indicated, all shares of the Company’s securities will be owned directly, and the person or entity listed as the beneficial owner has sole voting and investment power. Unless otherwise indicated, the address of all listed shareholders is 1920 Yonge Street, Suite 200, Toronto, Ontario, M4S 3E2.
| Name and Position of Beneficial Owner | Amount
and Nature of Beneficial Ownership(1) |
Percent
of Class |
||||||
| Directors and Executive Officers | ||||||||
| Eugene Williams, Chairman & Chief Executive Officer | 16,613,838 | (2) | 3.77 | % | ||||
| Daniel Geffken, Chief Financial Officer | 1,163,638 | (3) | * | |||||
| Elliot Goldstein, Former CEO & President | 15,830,421 | (4) | 3.60 | % | ||||
| Neil Cashman, Chief Scientific Officer & Director | 13,948,768 | (5) | 3.17 | % | ||||
| Madge “Maggie” K. Shafmaster, Director | 375,000 | (6) | * | |||||
| William Wyman, Director | 5,415,831 | (7) | 1.25 | % | ||||
| Patrick Kirwin, Director | 5,039,750 | (8) | 1.16 | % | ||||
| Richard Gregory, Director | 1,000,000 | (9) | * | |||||
| Josh Mandel-Brehm, Director | 312,500 | (10) | * | |||||
| Neil K. Warma, Director | 500,0000 | (11) | * | |||||
| All directors and executive officers as a group (10 people) | 45,827,658 | (12) | 10.06 | % | ||||
| >5% Shareholders | ||||||||
| Title 19 Investments LLC | 45,468,750 | (13) | 9.78 | % | ||||
| Crocker Mountain LLC | 34,562,500 | (14) | 7.75 | % |
* Represents less than 1%
Notes:
| (1) | For purposes of this table, beneficial ownership has been determined in accordance with the provisions of Rule 13d-3 of the Exchange Act, under which, in general, a person is deemed to be the beneficial owner of a security if he or she has or shares the power to vote or direct the voting of the security or the power to dispose of or direct the disposition of the security, or if he or she has the right to acquire beneficial ownership of the security within 60 days (“presently exercisable ”). Except as otherwise indicated, each director or executive officer has sole voting and investment power with respect to the shares shown, and none of such shares are pledged. |
97
| (2) | Includes 8,136,083 Common Shares underlying options and 473,939 Common Shares underlying warrants. | |
| (3) | Includes 750,000 Common Shares underlying options and 62,500 Common Shares underlying warrants, which are held by Danforth Advisors LLC. | |
| (4) | Includes 7,948,583 Common Shares underlying options and 473,939 Common Shares underlying warrants. | |
| (5) | Includes 7,136,083 Common Shares underlying options, 708,333 Common Shares underlying warrants and 63,708 Common Shares underlying DSUs. Also includes 10,000 common shares held by Rosemary Cashman, his spouse. | |
| (6) | Represents Common Shares underlying options. | |
| (7) | Includes 1,200,000 Common Shares underlying options and 439,582 Common Shares underlying warrants. | |
| (8) | Includes 200,000 Common Shares underlying warrants, 1,000,000 Common Shares underlying options, 939,900 Common Shares held by Patrick D. Kirwin Professional Corporation and 143,000 Common Shares held by Patrick Kirwin in a Tax Free Savings Account. Mr. Kirwin exercises the power to vote or direct the voting or the power to dispose or direct disposition of such securities. Also includes 343,000 Common Shares and 40,000 Common Shares underlying warrants held by Mrs. Jeananne Kirwin, Mr. Kirwin’s spouse. | |
| (9) | Represents Common Shares underlying options. | |
| (10) | Represents Common Shares underlying options. | |
| (11) | Represents Common Shares underlying options. | |
| (12) | Includes 1,093,750 Common Shares underlying options | |
| (13) | Includes 30,000,000 Common Shares underlying Debentures and 15,468,750 Common Shares underlying units, which units include 12,375,000 Common Shares and 3,093,750 Common Shares underlying warrants. Michael Gordon has sole voting and dispositive power over the Common Shares held by Title 19 Investments LLC. The address of Title 19 Investments LLC is c/o JDJFOS, P.O. Box 962049, Boston, MA 02196. | |
| (14) | Includes 9,000,000 Common Shares underlying Debentures and 5,112,500 Common Shares underlying warrants. Jeremy Sclar has sole voting and dispositive power over the Common Shares held by Crocker Mountain LLC. The address of Crocker Mountain LLC is 33 Boylston Street, Ste. 3000, Chestnut Hill, MA 02467. |
ITEM 5. DIRECTORS AND EXECUTIVE OFFICERS
The following table sets forth the individuals who we anticipate will be the directors and executive officers of the Company as of the filing of this Registration Statement and their respective positions.
| Name | Age | Position | ||||
| Eugene Williams | 62 | Chairman & Chief Executive Officer | ||||
| Daniel Geffken | 65 | Chief Financial Officer | ||||
| Gavin Malenfant | 60 | Chief Operating Officer | ||||
| Neil Cashman | 70 | Chief Scientific Officer & Director | ||||
| Maggie Shafmaster | 63 | Director | ||||
| William Wyman | 84 | Director | ||||
| Patrick Kirwin | 65 | Director | ||||
| Richard Gregory | 64 | Director | ||||
| Josh Mandel-Brehm | 39 | Director | ||||
| Neil Warma | 59 | Director | ||||
98
Director and Executive Officer Biographies
Eugene Williams, Chairman & CEO
Mr. Williams has served as Chairman and CEO of the Company since October 2021. Prior thereto, Mr. Williams served as Executive Chairman of the Company since July 2015. Prior thereto, Mr. Williams served as Chairman and Chief Executive Officer of Akashi (f/k/a DART Therapeutics, Inc.) from June 2010 to January 2014. Previously Mr. Williams was a senior executive at Genzyme, where he had broad management responsibilities in drug development, commercialization, and licensing.
Mr. Williams graduated from Harvard College with an Artium Baccalaureus degree in Economics and earned a Master of Business Administration from Harvard Business School.
Daniel Geffken, CFO
Mr. Geffken has served as CFO of the Company since March 2017. He is a co-founder of Danforth Advisors LLC since June 2011, and has served as Managing Director. Mr. Geffken has served on the board of directors of a number of public companies, including Windtree Therapeutics, Inc. since 2019, Arcturus Therapeutics, Inc. from November 2017 to May 2018, and Alcobra Pharmaceuticals Inc. from May 2013 to November 2017. Mr. Geffken has served on the board of directors of Elicio Therapeutics, a private company, since 2017.
Mr. Geffken earned a Bachelor of Science degree from the University of Pennsylvania and a Master of Business Administration from Harvard Business School.
Gavin Malenfant, COO
Mr. Malenfant has served as Chief Operating Officer of the Company since October 2021. Prior to joining ProMIS, Mr. Malenfant operated his own consulting business for series A companies. Mr. Malenfant’s experience is backed by nearly twenty-years with Genzyme, leading the rare disease program management organization and head of operations for research and development.
Mr. Malenfant earned a Bachelor of Science degree in Biology from the University of Massachusetts at Boston.
Neil Cashman, CSO & Director
Dr. Cashman has served as CSO and as a director of the Company since May 2004 and June 2010, respectively. Dr. Cashman has served as a Professor at the University of British Columbia (UBC) since July 2005 and will become Professor Emeritus as of February 1, 2022. He has also served as the Canada Research Chair in Neurodegeneration and Protein Misfolding Diseases at UBC from 2005 to 2019. He is also director of the ALS Clinic at Vancouver General Hospital since July 2005.
Dr. Cashman earned a Bachelor of Arts degree in Physics from Bowdoin College and a Medical Degree from the University of Massachusetts Medical School. Dr. Cashman served his residency in neurology with the University of Chicago Hospitals & Clinics.
Madge “Maggie” Shafmaster, Director
Dr. Shafmaster has served as a director of the Company since September 2021. Dr. Shafmaster has over 25 years of experience providing intellectual property advice to the biotechnology and pharmaceutical industries. Since 2014, Dr. Shafmaster has served as an independent intellectual property consultant to the biotech and pharma industries. Prior to this, she served from 2011 to 2014 as Vice President, Chief Patent Counsel for Sanofi Pasteur and from 2007 to 2011 as Senior Vice President, Chief Patent Counsel for Genzyme Corporation.
99
Dr. Shafmaster earned her Ph.D. in Molecular Biology and Virology from Cornell University Graduate School of Medical Sciences, a Juris Doctor from New York Law School and a Bachelor of Arts in Biology from the University of California Santa Cruz.
William Wyman, Director
Mr. Wyman has served as a director of the Company since March 2014. In 1984, Mr. Wyman co-founded Oliver Wyman & Co., a general management consulting firm. Since his retirement from the firm in 1995, Mr. Wyman has served as a director and advisor to nearly two dozen public and private companies in the finance and technology industries. Mr. Wyman has also served as a consultant and owner of Wyman Consulting Associates since 2016.
Mr. Wyman has been a member of the board of trustees of Dartmouth Hitchcock Medical Center, Mary Hitchcock Hospital and the Dartmouth Hitchcock Clinic, and currently serves on the Board Joint Development Committee. He is currently a member of the Board of Trustees of New England College. He served as a director of Allston Trading, LLC, a trading firm, since 2008, and as a member of the board of advisors of several private equity firms since 1995. He has also served on the National Academy of Sciences' committee on health equity.
Mr. Wyman earned his Bachelor of Arts degree in Economics from Colgate University and his Master of Business Administration from the Harvard Business School.
Patrick Kirwin, Director
Mr. Kirwin has served as a director of the Company since June 2015. Mr. Kirwin is senior partner at the law firm Kirwin LLP. Mr. Kirwin earned a Bachelor of Arts degree in Economics from the University of Alberta and a Juris Doctor from the University of Toronto Law School.
Richard Gregory, Director
Dr. Gregory has served as a director of the Company since October 2016. He has served as Executive Vice President and Chief Scientific Officer of ImmunoGen, Inc. from 2015 to 2019. Dr. Gregory has been a Fellow of the American Institute for Medical and Biological Engineering since 2015. He has served on the board of directors of Homology Medicines, Inc. and Cambridge Therapeutic Technologies since 2015 and March 2021, respectively.
Dr. Gregory earned a Bachelor of Science degree in Biochemistry from Virginia Polytechnic Institute and State University (Virginia Tech) and a Ph.D. in Biochemistry from the University of Massachusetts Amherst.
Josh Mandel-Brehm, Director
Mr. Mandel-Brehm has served as a director of the Company since September 2021. Mr. Mandel-Brehm has served as President and Chief Executive Officer of CAMP4 Therapeutics Corporation since May 2017 and as entrepreneur partner with Polaris Partners. Prior to May 2017, Mr. Mandel-Brehm served in business development for Biogen Corporation from May 2013 to May 2017. He has also been a founder and board member for Vico Therapeutics B.V. since October 2019.
Mr. Mandel-Brehm earned a Bachelor of Arts degree in Biology from Washington University in St. Louis and a Master of Business Administration from the University of Michigan.
100
Neil K. Warma, Director
Mr. Warma has served as a director of the Company since May 2021. Mr. Warma has been a healthcare entrepreneur for over 25 years having managed and advised numerous biotechnology and pharmaceutical companies across the globe. Mr. Warma has served as the General Manager of I-Mab Biopharma U.S., a publicly-traded global biopharmaceutical company since September 2019. Mr. Warma was founder and from 2018 to 2019 served as CEO of Biohealth Care, LLC, which provided advisory services to the healthcare industry. Previously, Mr. Warma was President and CEO and a member of the board of directors of Opexa Therapeutics, Inc., a publicly-traded biopharmaceutical company from 2008 to 2017. He was President, CEO and Director of Viron Therapeutics from 2004 to 2007 and prior to that held several senior positions at Novartis AG in Basel, Switzerland.
Mr. Warma has served as a director for Genexine Ltd., a public company, and Biotechnology Innovation Organization since March 2021 and November 2020, respectively.
Mr. Warma earned a Bachelor of Science degree in Neuroscience from the University of Toronto and a Master of Business Administration from York University.
Significant Employee
Johanne Kaplan
Dr. Kaplan has served as the Company’s Chief Development Officer (“CDO”) since 2016, assuming the role in a full-time capacity on January 1, 2022. Prior to taking on the CDO role full-time, Dr. Kaplan also served as Chief Scientific Officer at Shepherd Therapeutics from 2016 to 2021 and as Chief Scientific Officer at Epiva Biosciences from 2015 to 2016. Before joining the Company, Dr. Kaplan held increasing positions of responsibility at Sanofi Genzyme, from 1992 through 2015, most recently serving as Vice President of Research at Sanofi Genzyme from 2005 until her retirement in 2015. As Vice President of Neuroimmunology Research, she led the contribution of the Genzyme science team supporting the approval of Lemtrada (alemtuzumab) and Aubagio (teriflunomide) for the treatment of relapsing-remitting multiple sclerosis. She also established partnerships for the development of novel therapies for neuroinflammatory disorders. Prior to joining Genzyme, Dr. Kaplan was an Associate Immunopathologist at SmithKline Beecham where she established an immunotoxicology program. Her work has resulted in over 70 scientific publications and multiple patents. Dr. Kaplan holds a PhD in Microbiology & Immunology from McGill University in Montreal, Canada and conducted post-doctoral studies at the Albert Einstein College of Medicine in New York.
While Dr. Kaplan does not serve in a policy-making function for the Company and is therefore not an executive officer for U.S. securities law purposes, her scientific background and experience play an important role in the Company’s operations
| Member | Independent | Audit | Corporate
Governance and Nominating |
Compensation |
| Eugene Williams | ||||
| Neil Cashman | ||||
| Maggie Shafmaster | ü | ü | ||
| William Wyman | ü | ü | ü | |
| Patrick Kirwin | ü | ü | ||
| Richard Gregory | ü | ü | ||
| Josh Mandel-Brehm | ü | ü | ||
| Neil Warma | ü | ü | ü | ü |
101
Audit Committee
The audit committee of the Board (the “Audit Committee”) assists the Company’s Board in fulfilling its oversight responsibilities relating to financial accounting and reporting process and internal controls for the Company and ensuring the adequacy and effectiveness of the Company’s risk management programs. The Audit Committee reviews the financial reports and other financial information provided by the Company to regulatory authorities and its shareholders, as well as reviews the Company’s system of internal controls regarding finance and accounting, including auditing, accounting and financial reporting processes.
Composition of the Audit Committee
As of the date of filing of this Registration Statement, the following are the members of the Audit Committee:
| Name of Member | Independent(1) | Financially Literate(2) | ||||||
| William Wyman | Yes | Yes | ||||||
| Patrick Kirwin | Yes | Yes | ||||||
| Neil Warma | Yes | Yes |
Notes:
| (1) | A member of the Audit Committee is independent if he or she has no direct or indirect ‘material relationship’ with the Company. A material relationship is a relationship which could, in the view of the Company’s Board, reasonably interfere with the exercise of a member’s independent judgment. Any executive officer of the Company is deemed to have a material relationship with the Company. |
| (2) | A member of the Audit Committee is financially literate if he or she has the ability to read and understand a set of financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Company’s financial statements. |
Relevant Education and Experience
Each member of the Audit Committee has experience relevant to his or her responsibilities as an Audit Committee member. See Item 5—“Directors and Executive Officers” for a description of the education and experience of each Audit Committee member.
Audit Committee Oversight
At no time since the commencement of the Company’s most recently completed financial year were any audit committee recommendations to nominate or compensate an external auditor not adopted by the Board.
Audit Committee Charter
The Board has adopted a written charter for the Audit Committee, which sets out the Audit Committee’s responsibilities. The Audit Committee performs a number of roles including (i) assisting directors to meet their oversight responsibilities, (ii) enhancing communication between directors and the external auditors; (iii) ensuring the independence of the external auditors; (iv) increasing the credibility and objectivity of financial reports; and (v) strengthening the role of the directors by facilitating in-depth discussions among directors, management and the external auditor. The Audit Committee has been delegated responsibility for: (i) the Company’s internal audit function; (ii) the integrity of our consolidated financial statements and accounting and financial processes and the audits of our consolidated financial statements; (iii) compliance with legal and regulatory requirements; (iv) the external auditors’ qualifications and independence; (v) the work and performance of financial management and external auditors; and (vi) the system of disclosure controls and procedures and system of internal controls regarding finance, accounting, legal compliance and risk management established by management and the Board. The Audit Committee has unrestricted access to all books and records of the Company and may request any information as it may deem appropriate. It also has the authority to retain and compensate special legal, accounting, financial and other consultants or experts in the performance of its duties.
102
Corporate Governance and Nominating Committee
The Corporate Governance and Nominating Committee of the Board assists the Board in fulfilling its oversight responsibilities relating to the corporate governance of the Company and the size, structure, and membership of the Board and its committees.
Composition of the Corporate Governance and Nominating Committee
As of the date of this Registration Statement, the following are the members of the Corporate Governance and Nominating Committee:
| Name of Member | Independent(1) | |||
| Josh Mandel-Brehm | Yes | |||
| Maggie Shafmaster | Yes | |||
| Neil Warma | Yes |
Notes:
(1) A member of the Corporate Governance and Nominating Committee is independent if he or she has no direct or indirect ‘material relationship’ with the Company. A material relationship is a relationship which could, in the view of the Company’s Board, reasonably interfere with the exercise of a member’s independent judgment. Any executive officer of the Company is deemed to have a material relationship with the Company
Corporate Governance and Nominating Committee Charter
The Board has adopted a written charter for the Corporate Governance and Nominating Committee, which sets out the Corporate Governance and Nominating Committee’s responsibilities. The Corporate Governance and Nominating Committee has been delegated responsibility for: i) reviewing the appropriate skills and characteristics required of Board members in the context of the current make-up of the Board; and ii) assess the Board’s compliance with laws and policies relating to the independence of certain Board members.
Compensation Committee
The Compensation Committee of the Board assists the Board in fulfilling its oversight responsibilities relating to the recruitment, compensation, evaluation and retention of senior management and other key employees, and in particular the CEO, with the skills and expertise needed to enable the Company to achieve its goals and strategies at competitive compensation and with appropriate performance incentives.
Composition of the Compensation Committee
As of the date of this Registration Statement, the following are the members of the Compensation Committee:
| Name of Member | Independent(1) | |||
| Richard Gregory | Yes | |||
| Neil Warma | Yes | |||
| William Wyman | Yes |
Notes:
(1) A member of the Compensation Committee is independent if he or she has no direct or indirect ‘material relationship’ with the Company. A material relationship is a relationship which could, in the view of the Company’s Board, reasonably interfere with the exercise of a member’s independent judgment. Any executive officer of the Company is deemed to have a material relationship with the Company.
Compensation Committee Charter
The Board has adopted a written charter for the Compensation Committee, which sets out the Compensation Committee’s responsibilities. The Compensation Committee has been delegated responsibility for reviewing: i) compensation policies and guidelines for supervisory and management personnel of the Company; ii) corporate benefits, bonuses and other incentives, including share options and restricted share awards; iii) corporate goals and objectives relevant to chief executive officer compensation; iv) non-chief executive officer and director compensation, incentive compensation plans and equity-based plans; v) the competitiveness and appropriateness of the Company’s policies relating to the compensation of executive officers; and vi) any material changes or trends in human resources policy, procedure, compensation and benefits.
103
We believe that each of the members of our Board has the experience, qualifications, attributes and skills that make him or her suitable to serve as our director, in light of our highly regulated cryptocurrency business, our complex operations and large number of employees. See Item 5—”Directors and Executive Officers” for a description of the education and experience of each director.
Eugene Williams’s specific qualifications, experience, skills and expertise include:
| · | Experience as Chairman and CEO of the Company and in other executive leadership capacities in the pharmaceutical and biotechnology industries.
| ||
| · | A deep understanding of entrepreneurship, of drug development and of the pharmaceutical and biotechnology industries. | ||
| · | Corporate strategy. |
Neil Cashman’s specific qualifications, experience, skills and expertise include:
| · | Pharmaceutical and biotechnology industry and academic expertise in protein misfolding, neurodegeneration, and neurological clinical care. | ||
| · | Experience as CSO. | ||
| · | Previous history on the Company’s Board. |
Maggie Shafmaster’s specific qualifications, experience, skills and expertise include:
| · | Experience providing intellectual property advice to the biotechnology and pharmaceutical industries. | ||
| · | Extensive business experience in various executive level roles. | ||
| · | Corporate strategy. |
William Wyman’s specific qualifications, experience, skills and expertise include:
| · | Previous history on the Company’s Board. | ||
| · | Extensive business experience in various executive and board level roles. | ||
| · | Operating and management experience. |
Patrick Kirwin’s specific qualifications, experience, skills and expertise include:
| · | Previous history on the Company’s Board. | ||
| · | Knowledge of past and current business strategies. |
104
Richard Gregory’s specific qualifications, experience, skills and expertise include:
| · | Extensive business experience in various executive and board level roles. | ||
| · | Pharmaceutical and biotechnology industry knowledge. | ||
| · | Corporate governance. |
Josh Mandel-Brehm’s specific qualifications, experience, skills and expertise include:
| · | Extensive business experience in various executive level roles in the biotechnology industry. | ||
| · | A deep understanding of entrepreneurship and of the biotechnology industry. | ||
| · | Corporate Strategy. |
Neil K. Warma’s specific qualifications, experience, skills and expertise include:
| · | Experience in executive level roles in the pharmaceutical and biotechnology industries. | ||
| · | Operating and management experience. | ||
| · | A deep understanding of entrepreneurship and of the pharmaceutical and biotechnology industries. |
The Board believes these qualifications bring a broad set of complementary experience to the Board’s discharge of its responsibilities.
Conflicts of Interest—Board Leadership Structure and Risk Oversight
Conflicts of interest may arise as a result of the directors and officers of the Company also holding positions as directors or officers of other companies. Some of the individuals that are directors and officers of the Company have been and will continue to be engaged in the identification and evaluation of assets, businesses and companies on their own behalf and on behalf of other companies, and situations may arise where the directors and officers of the Company will be in direct competition with the Company. Conflicts, if any, will be subject to the procedures and remedies provided under the Company’s Code of Business Conduct and Ethics.
ITEM 6. EXECUTIVE COMPENSATION
The following discussion describes the significant elements of the compensation of each person who served as the Company’s Chief Executive Officer (“CEO”) during 2021 and two most highly compensated executive officers (collectively, the “named executive officers” or “NEOs”). As at December 31, 2021, the NEOs of the Company were Eugene Williams (Executive Chairman & CEO), Elliot Goldstein (former CEO and President), Daniel Geffken (CFO) and Neil Cashman (CSO).
The Company’s policy with respect to compensation of the named executive officers and other officers of the Company is based upon the principles that total compensation must: (1) be competitive in order to help attract and retain the talent needed to lead and grow the Company’s business; (2) provide a strong incentive for executives and key employees to work towards the achievement of the Company’s goals; and (3) ensure that the interests of management and the Company’s shareholders are aligned.
105
When determining the compensation of its executive officers, the Compensation Committee considers: (i) recruiting and retaining executives critical to the success of the Company and the enhancement of shareholder value; (ii) providing fair and competitive compensation compared to the remuneration paid by other reporting issuers similarly placed within the same business as the Company; (iii) balancing the interests of management and the Company’s shareholders; and (iv) rewarding performance, both on an individual basis and with respect to operations in general. In order to achieve these objectives, the compensation paid to the Company’s executive officers consists of two components: (i) base salary; and (ii) long-term equity incentives in the form of share options. In making compensation determinations, external sources are consulted when deemed necessary by the Compensation Committee. The members of the Compensation Committee are disclosed under Item 5 of this Registration Statement.
The total compensation paid to each of the named executive officers of the Company consists of a base salary or consulting fee and share options to reward and retain NEOs. Total compensation paid to each NEO reflects the executive’s overall experience, responsibility and time committed to the organization. The goal of the Company is to pay base salary compensation to retain the NEOs in the range of industry peers, while maintaining the overall goal that total compensation should include variable and long-term components as well.
With respect to the CEO’s compensation in particular, the CEO’s base salary is determined after considering the salary levels of other executives with similar responsibilities and experience. The CEO’s base salary is compared to salary levels of comparable executives at a variety of companies, with particular emphasis on biotechnology companies with similar market capitalizations.
Options are granted by the Board to employees, executive officers, including the named executive officers, and directors pursuant to the Company’s Stock Option Plan. The purpose of the Stock Option Plan is to attract, retain and motivate these individuals and create incentives for them to contribute toward the long-term goals of the Company. Moreover, the Stock Option Plan aims to align the interests of participants with the Company’s Shareholders through opportunities of increased equity-based ownership in the Company.
The Board may also grant DSUs to senior officers, including any named executive officers, under the Company’s DSU Plan, which provides an alternative form of compensation to satisfy annual and special bonuses payable to senior officers. The number of DSUs granted is determined by dividing the applicable bonus amount by the fair market value of the Common Shares as at the last trading day before calculation in accordance with TSX policies. Recipients of DSUs cannot exercise their DSUs until such time as they cease to be a senior officer at which time they may elect to receive one Common Share for each whole DSU they hold at the time they cease to be eligible to participate in the DSU Share Unit Plan.
Approach to Risk
The Board understands that compensation practices can have unintended risk consequences. The Compensation Committee continually reviews the Company’s compensation policies to identify any practice that might encourage an employee to expose the Company to unacceptable risk. At the present time, the Compensation Committee is satisfied that the current executive compensation program does not encourage the Company’s executive officers, including the NEOs, to expose the Company to inappropriate risk. The Board takes a conservative approach to executive compensation, rewarding individuals for the success of the Company once that success has been demonstrated and encouraging them to continue that success through the grant of long-term incentive awards.
106
Hedging Policy
There are no specific requirements to prevent an NEO or director from purchasing financial instruments including, for greater certainty, prepaid variable forward contracts, equity swaps, collars, or units of exchange funds that are designed to hedge or offset a decrease in market value of equity securities granted as compensation or held, directly or indirectly, by the NEO or director.
Summary Compensation Table for 2021
The following table sets forth all compensation paid to or earned by the named executive officers of the Company in the last fiscal year.
| Name
and Principal Position | Year | Salary ($)(1) | Option Awards ($)(2) | All
Other Compensation ($)(3) | Total
($) | ||||||||||||||
| Eugene
Williams Executive Chairman & CEO | 2021 | $ | 360,000 | $ | — | $ | 21,057 | $ | 381,057 | ||||||||||
Elliot Goldstein Former CEO & President(4) | 2021 | $ | 50,000 | $ | — | $ | 12,677 | $ | 62,677 | ||||||||||
| Daniel Geffken Chief Financial Officer | 2021 | $ | — | $ | 71,670 | $ | 97,200 | $ | 168,870 | ||||||||||
| Neil Cashman Chief Scientific Officer | 2021 | $ | 79,017 | $ | — | $ | — | $ | 79,017 | ||||||||||
(1) The amounts reported in the Salary column reflect payments made pursuant to various consulting agreements. In addition, during 2021 the named executive officers were paid salary that had been accrued for service in prior years but not paid in those prior years, as follows: Eugene Williams, $257,677; Elliot Goldstein, $257,677.
(2) The amounts reported in the Option Awards column reflects aggregate grant date fair value computed in accordance with ASC Topic 718, Compensation—Stock Compensation. These amounts reflect our calculation of the value of these awards at the grant date and do not necessarily correspond to the actual value that may ultimately be realized by the named executive officer. Please refer to Note 13 of the Notes to the Consolidated Financial Statement for additional information regarding share based compensation. Option Awards granted in 2021 with respect to Mr. Geffken’s service were granted to Danforth Advisors, LLC. Mr. Geffken serves as Managing Director of such entity.
(3) Amounts reported in the All Other Compensation column reflect payments made to Mr. Williams and Dr. Goldstein for health insurance costs. The amount reported for Mr. Geffken reflects payments made to Danforth Advisors, LLC as a retainer for Mr. Geffken’s services.
(4) Dr. Goldstein ceased serving as CEO of the Company on June 30, 2021 and ceased serving as President and a director on December 31, 2021.
107
Consulting and Employment Agreements
Eugene Williams. The Chairman and CEO services provided by Mr. Williams to the Company were historically provided pursuant to a consulting agreement entered into between the Company and Virtua, LLC dated June 29, 2015 (the “Virtua Consulting Agreement”). Pursuant to the terms of the Virtua Consulting Agreement, Mr. Williams was appointed Executive Chair of the Company beginning on the date of the agreement and continuing until either party provides notice of its intent to terminate the agreement for any reason, at any time, upon 30 days’ written notice, which was able to be waived by either party, upon 15 days written notice in the event of a breach by either party, or on the written agreement of both parties. Subject to adjustment by the Board, the Company agreed to pay Virtua, LLC a $30,000 fixed fee per month, with $10,000 of that monthly fee to be allocated for services provided by Mr. Williams, plus reimbursement for reasonable expenses. The Virtua Consulting Agreement also provided for the grant of options to Mr. Williams under the Company’s Stock Option Plan equal to five percent of the shares issued and outstanding immediately following the completion or termination of the private placement offering announced by the Company on May 22, 2015. Such options expire 10 years following their grant date and entitle Mr. Williams to acquire shares at the market price on the grant date, with one quarter of such options immediately vesting and the balance vesting in equal installments on the last day of each month for 36 months, except, in the event of a change of control or in the case where there is a termination without good reason, on the occurrence of which the entire balance shall vest immediately. In the event the Virtua Consulting Agreement is terminated other than for a change of control or where there is a termination without good reason, unvested options were to cease vesting as of such termination date. The foregoing description is qualified in its entirety by reference to the Virtua Consulting Agreement, which is included as Exhibit 10.11 hereto and incorporated by reference herein.
On December 21, 2021, the Company extended to Mr. Williams an offer of employment (the “Williams Offer Letter”) to serve as the Company’s CEO beginning January 1, 2022. Pursuant to the terms of the of the Williams Offer, Mr. Williams’ compensation for service as the Company’s CEO is set at $480,000 and Mr. Williams is eligible to participate in any and all bonus and benefit programs that the Company makes available to its employees. In addition, Mr. Williams was awarded 3,000,000 share options on February 10, 2022 priced at $0.14, vesting 1/48th monthly over a four-year period, with the options expiring on February 10, 2032. Upon termination, all vested options will be exercisable at any time during the twelve months following termination. In the event of termination of Mr. Williams’ employment by the Company without cause, by Mr. Williams with good reason, or termination by way of a change in control, then upon Mr. Williams’ execution of a release of claims, Mr. Williams is entitled to an aggregate amount equivalent to eighteen months of his then-current base salary The Williams Offer Letter supersedes all prior agreements regarding Mr. Williams’ services to and compensation from the Company, including the Williams Consulting Agreement. The foregoing description is qualified in its entirety by reference to the Williams Offer Letter, which is included as Exhibit 10.36.
Elliot Goldstein. Dr. Goldstein was party to a consulting agreement with the Company dated April 1, 2021 for CEO consulting services (the “Goldstein Consulting Agreement”). Under the Goldstein Consulting Agreement, Dr. Goldstein agreed to provide the Company CEO consulting services for a six month period in exchange for a monthly payment of $5,000, subject to increases as approved by the Board. Dr. Goldstein entered into a second consulting agreement dated October 1, 2021 for President consulting services (the “Second Goldstein Consulting Agreement”, and together with the Goldstein Consulting Agreement, the “Goldstein Consulting Agreements”). Under the Second Goldstein Consulting Agreement, Dr. Goldstein agreed to provide the Company President consulting services until September 30, 2022 in exchange for a monthly payment of $10,000 plus reasonable expenses incurred in connection with such services. Prior to entry into the Goldstein Consulting Agreements, Mr. Goldstein provided services to the Company under the Virtua Consulting Agreement. Pursuant to the terms of the Virtua Consulting Agreement, Mr. Goldstein was appointed Chief Executive Officer of the Company beginning on the date of the agreement and continuing until either party provides notice of its intent to terminate the agreement for any reason, at any time, upon 30 days’ written notice, which was able to be waived by either party, upon 15 days written notice in the event of a breach by either party, or on the written agreement of both parties. The Company agreed to pay Virtua, LLC a $30,000 fixed fee per month, with $20,000 of that monthly fee to be allocated for services provided by Mr. Goldstein, plus reimbursement for reasonable expenses. The foregoing descriptions are qualified in their entirety by reference to the Goldstein Consulting Agreement, the Second Goldstein Consulting Agreement and Virtua Consulting Agreement, which are included as Exhibits 10.8, 10.8.1 and 10.11 hereto and incorporated by reference herein.
108
Daniel Geffken. The CFO services provided by Mr. Geffken are provided pursuant to a consulting agreement entered into between the Company and Danforth Advisors, LLC dated October 17, 2016, and as amended from time-to-time (the “Danforth Consulting Agreement”). Under the Danforth Consulting Agreement, Mr. Geffken agreed to provide the Company the customary services of a CFO at an hourly rate of $325 for a one year term. On March 27, 2017, the Danforth Consulting Agreement was amended to provide for services based on a $5,000 monthly retainer, subject to a 4% annual increase, plus expenses. The Danforth Consulting Agreement was subsequently amended on December 12, 2017 to extend the term for an additional year and on August 31, 2018 to extend the term for an additional year. The Danforth Consulting Agreement was further amended on November 10, 2021 to extend the term of the consulting agreement through October 29, 2024 and to set Mr. Geffken’s compensation at a fixed monthly fee of $15,000. The Danforth Consulting Agreement provides for an extension of terms by the mutual agreement of the parties and that either party may terminate the agreement upon sixty days prior written notice to the other, or 30 days in the case of termination for cause. The Corporation also issued 500,000 stock options to Danforth Advisors, LLC, which will vest as follows: 25% vested immediately upon the grant of options and the balance will vest in equal installments over 36 months. The foregoing description is qualified in its entirety by reference to the Danforth Consulting Agreement and the amendments thereto, which are included as Exhibits 10.12 and 10.12.1, 10.12.2, 10.12.3 and 10.12.4 hereto and incorporated by reference herein.
Neil Cashman. Dr. Cashman is party to a consulting and advisory agreement with the Company dated March 1, 2005 (the “Cashman Consulting Agreement”) for CSO consulting services. The Cashman Consulting Agreement provides that it shall remain in effect until terminated by either party, with the Company agreeing to provide Dr. Cashman six month’s written notice and Dr. Cashman agreeing to provide the Company thirty day’s written notice. In return for the CSO services, the Company agreed to pay Dr. Cashman a monthly consulting fee of $5,000, plus expenses, subject to adjustment as approved by the Board. Effective March 1, 2017, the monthly consulting fee payable to the CSO was increased to CDN$9,000 per month pursuant to a Board authorized resolution. The foregoing description is qualified in its entirety by reference to the Cashman Consulting Agreement, which is included as Exhibit 10.10 hereto and incorporated by reference herein.
Outstanding Equity Awards Table for 2021
The following table sets forth outstanding equity awards for the named executive officers of the Company at fiscal 2021 year end.
| Option Awards | Stock Awards(1) | |||||||||||||||||||||||||||
| Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities Underlying Unexercised Options (#) Unexercisable |
Equity
Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) |
Option Exercise Price ($)(2) |
Option Expiration Date |
Number
of shares or units of stock that have not vested (#) |
Market value of shares or units of stock that have not vested (US$)(3) |
Equity incentive plan awards: number of unearned shares, units or other rights that have not vested (#) |
Equity incentive plan awards: market or payout value of unearned shares, units or other rights that have not vested ($) |
||||||||||||||||||||
| Eugene Williams | 4,729,300 Common Shares | $ | 0.0319 | (4) | 7/6/2025 | |||||||||||||||||||||||
| 2,219,283 Common Shares | $ | 0.0513 | (5) | 7/31/2025 | ||||||||||||||||||||||||
| 1,000,000 Common Shares | $ | 0.3707 | (6) | 3/29/2028 | ||||||||||||||||||||||||
| Elliot Goldstein | 4,729,300 Common Shares | $ | 0.0319 | (4) | 7/6/2025 | |||||||||||||||||||||||
| 2,219,283 Common Shares | $ | 0.0513 | (5) | 7/31/2025 | ||||||||||||||||||||||||
| 1,000,000 Common Shares | $ | 0.3707 | (6) | 3/29/2028 | ||||||||||||||||||||||||
| Daniel Geffken(7) | 500,000 Common Shares | $ | 0.14198 | (8) | 3/1/2027 | |||||||||||||||||||||||
| 500,000 Common Shares | 458,333 Common Shares | $ | 0.14198 | (9) | 11/12/2031 | |||||||||||||||||||||||
| Neil Cashman | 4,729,300 Common Shares | $ | 0.0319 | (4) | 7/6/2025 | 15,484 | $ | 1,548 | ||||||||||||||||||||
| 2,219,283 Common Shares | $ | 0.0513 | (5) | 7/31/2025 | 19,938 | $ | 1,994 | |||||||||||||||||||||
| 28,286 | $ | 2,829 | ||||||||||||||||||||||||||
109
(1) The Company’s only share-based awards are DSUs (other than options) that have been granted under the DSU Plan. DSUs only vest in full upon separation from service.
(2) Pursuant to the Company’s Stock Option Plan, the option exercise price is granted in Canadian dollars. This presentation has been converted into U.S. dollars using the Bank of Canada daily exchange rate for December 31, 2021, which was US$1.00 to C$1.2678.
(3) The value of the unvested share-based awards was calculated based on the closing price of the Company’s Common Shares on the TSX on December 30, 2021, which was C$0.13 (US$0.10). The Bank of Canada exchange rate as of December 30, 2021 was US$1.00 to C$1.2678.
(4) The option was granted on July 6, 2015 with an exercise price of C$0.0405. The option vested ¼ immediately with balance having vested ratably over 36 months.
(5) The option was granted on July 31, 2015 with an exercise price of C$0.0650. The option vested ¼ immediately with balance having vested ratably over 36 months.
(6) The option was granted on March 29, 2018 with an exercise price of C$0.364. The option vested ¼ immediately with balance having vested ratably over 36 months.
(7) Options are held by Danforth Advisors, LLC. Mr. Geffken serves as Managing Director of such entity.
(8) The option was granted on March 1, 2017 with an exercise price of C$0.18. The option vested 1/5 immediately with balance having vested ratably over 36 months.
(9) The option was granted on November 12, 2021 with an exercise price of C$0.18. The option vests ratably over 12 months.
Retirement Benefit Plans
The Company does not have any retirement benefit plans.
Termination and Change in Control Benefits
The Company does not offer a formal plan providing for any termination or change in control benefits. For information about change in control benefits for Messrs. Williams and Malenfant, see “Consulting and Employment Agreements” above.
DIRECTOR COMPENSATION
As of December 31, 2021, the Company had eight directors, two of whom were also employees: Eugene Williams (Chairman and CEO) and Neil Cashman (CSO). The remaining six directors were considered independent directors at such time, namely Richard Gregory, Patrick Kirwin, Josh Mandel-Brehm, Maggie Shafmaster, Neil K. Warma and William Wyman.
Directors who hold positions as executive officers with the Company do not receive additional compensation for their service as directors. Mr. Williams and Dr. Cashman did not receive any additional compensation for their services as directors during the year ended December 31, 2021. For a description of the compensation paid to Mr. Williams and Dr. Cashman, see “Summary Compensation Table for 2021,” above.
Each member of the Company’s Board is entitled to reimbursement for reasonable travel and other expenses incurred in connection with attending Board meetings and meetings for any committee on which he or she serves.
110
Compensation of Directors
The form and amount of director compensation is reviewed annually and as deemed advisable by the Compensation Committee, which shall make recommendations to the Board based on such review. The Compensation Committee reviews director compensation on an annual basis to ensure that the Company offers director compensation that is: (i) commensurate with the efforts the Company expects from existing Board members; (ii) competitive in the Company’s industry in order that the Company might attract the best possible candidates to assist the Company and its shareholders in a fiduciary capacity; and (iii) aligned with shareholder interests as the Company grows. The Board retains the ultimate authority to determine the form and amount of director compensation.
Director Compensation for 2021
The following table sets forth all compensation paid to or earned by each director of the Company during fiscal year 2021.
| Name(1) | Fees Earned or | Option ($)(3) | Total ($) | |||||||||
| Richard Gregory | $ | 30,000 | $ | 67,269 | $ | 107,269 | ||||||
| Patrick Kirwin | $ | 30,000 | $ | 67,269 | $ | 107,269 | ||||||
| Josh Mandel-Brehm | $ | 13,333 | $ | 83,297 | $ | 96,630 | ||||||
| Maggie Shafmaster | $ | 10,000 | $ | 51,182 | $ | 61,182 | ||||||
| Neil Warma | $ | 25,000 | $ | 74,302 | $ | 99,302 | ||||||
| William Wyman | $ | 30,000 | $ | 67,269 | $ | 107,269 | ||||||
| Johannes Roth(4) | $ | -- | $ | -- | $ | -- | ||||||
(1) Mr. Williams, Dr. Cashman and Dr. Goldstein, who served as executive officers during 2021, did not receive any compensation for their Board service.
(2) Cash fees paid to non-employee directors.
(3) The amounts reported in the Option Awards column reflects aggregate grant date fair value computed in accordance with ASC Topic 718, Compensation—Stock Compensation. These amounts reflect our calculation of the value of these awards at the grant date and do not necessarily correspond to the actual value that may ultimately be realized by the director. Please refer to Note 13 of the Notes to the Consolidated Financial Statement for additional information regarding share based compensation.
(4) Mr. Roth resigned from the Board on February 1, 2021 and did not receive any compensation for his service in 2021.
Compensation Committee Interlocks and Insider Participation
See Item 7 — “Certain Relationships and Related Transactions and Director Independence” for further details.
None of the Company’s executive officers served as a member of the compensation committee (or other Board committee performing equivalent functions or, in the absence of any such committee, the entire Board) of another entity, one of whose executive officers served as a director of the Company or on the Compensation Committee, during the fiscal year ended December 31, 2021. None of the Company’s executive officers served as a director of another entity, one of whose executive officers served on the Compensation Committee, during the fiscal year ended December 31, 2021.
111
ITEM
7. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND
DIRECTOR INDEPENDENCE
For the Company, a related party transaction includes any transaction or proposed transaction in which:
| · | the Company is or will be a participant; |
| · | the aggregate amount involved exceeds $17,000 (approximately 1% of the Company’s average assets for the last two fiscal years) in any fiscal year; and |
| · | any related party has or will have a direct or indirect material interest. |
Related parties include any person who is or was (since the beginning of the last fiscal year, even if such person does not presently serve in that role) an executive officer or director of the Company, any shareholder beneficially owning more than 5% of any class of the Company’s voting securities or an immediate family member of any such persons. Immediate family member means any child, stepchild, parent, stepparent, spouse, sibling, mother-in-law, father-in-law, son-in-law, daughter-in-law, brother-in-law, sister-in-law, and any person (other than a tenant or employee) sharing the household of such person.
The Audit Committee is charged with oversight over related party transactions entered into by the Company.
112
Company Transactions with Related Parties
The Company has entered into related party transactions as follows:
Neil Cashman. Dr. Cashman is a Professor of Neurology with UBC. Please see Item 1 — “Business” for more information regarding the Company’s relationship with UBC.
Eugene Williams. Payments for Mr. William’s services were pursuant to a consulting agreement with Virtua, LLC. Virtua, LLC’s sole line of business was providing management services to the Company. Details regarding the consulting agreement are set forth in Item 6 – “Executive Compensation.”
Daniel Geffken. Mr. Geffken serves as Managing Director of Danforth Advisors, LLC. Payments for CFO services provided to the Company by Mr. Geffken are made to Danforth Advisors, LLC. For the years ended 2021, 2020 and 2019, the Company paid $326,619.75, $232,083.40 and $227,755.74, respectively, to Danforth Advisors, LLC for services provided pursuant to a consulting agreement. Additionally, on March 1, 2017 and November 12, 2021, the Company granted 500,000 and 500,000 share options, respectively, to Danforth Advisors, LLC pursuant to the same agreement. For additional information regarding this agreement and the amounts paid for Mr. Geffken’s services, see Item 6 – “Executive Compensation.”
Elliot Goldstein. Dr. Goldstein previously served as CEO and President of the Company. Payments for Dr. Goldstein’s services were pursuant to a consulting agreement with Virtua, LLC. Virtua, LLC’s sole line of business was providing management services to the Company. Details regarding the consulting agreement are set forth in Item 6 – “Executive Compensation.”
Gavin Malenfant. Mr. Malenfant was paid $41,300 as a consultant in 2021.
Please see Item 6 — “Executive Compensation” for details regarding executive and director compensation.
For purposes of this Registration Statement, the independence of our directors is determined under the corporate governance rules of the Nasdaq. The independence rules of Nasdaq include a series of objective tests, including that an “independent” person will not be employed by us and will not be engaged in various types of business dealings with us. In addition, the Board is required to make a subjective determination as to each person that no material relationship exists with the Company either directly or as a partner, shareholder or officer of an organization that has a relationship with the Company. It has been determined by the Board that six of our directors that we expect to be on the Board as of the Effective Date are independent persons under the independence rules of the Nasdaq: Richard Gregory, Patrick Kirwin, Josh Mandel-Brehm, Maggie Shafmaster, Neil K. Warma and William Wyman.
113
At present, to the Company’s knowledge, there are no material pending legal proceedings or regulatory actions to which the Company is or was a party to or of which any of its property is or was the subject of, and no such proceedings or actions are known by the Company to be contemplated.
ITEM
9. MARKET PRICE OF AND DIVIDENDS ON THE REGISTRANT’S COMMON EQUITY
AND RELATED SHAREHOLDER MATTERS
The Common Shares of the Company are traded on the TSX under the symbol “PMN.” The following table sets forth trading information for the Common Shares for the periods indicated, as quoted on the TSX:
| Period | Low Trading Price (C$) | High
Trading Price (C$) | ||||||
| Year Ending December 31, 2022 | ||||||||
| First Quarter (through March 31, 2022) | $ | 0.13 | $ | 0.14 | ||||
| Year Ending December 31, 2021 | ||||||||
| Fourth Quarter (December 30, 2021) | $ | 0.12 | $ | 0.20 | ||||
| Third Quarter (September 30, 2021) | $ | 0.18 | $ | 0.27 | ||||
| Second Quarter (June 30, 2021) | $ | 0.17 | $ | 0.27 | ||||
| First Quarter (March 31, 2021) | $ | 0.08 | $ | 0.22 | ||||
| Year Ending December 31, 2020 | ||||||||
| Fourth Quarter (December 31, 2020) | $ | 0.08 | $ | 0.17 | ||||
| Third Quarter (September 30, 2020) | $ | 0.13 | $ | 0.24 | ||||
| Second Quarter (June 30, 2020) | $ | 0.12 | $ | 0.31 | ||||
| First Quarter (March 31, 2020) | $ | 0.10 | $ | 0.23 | ||||
The Common Shares of the Company are also traded on the OTCQB Venture Market under the symbol “ARFXF.” The following table sets forth trading information for the Common Shares for the periods indicated, as quoted on the OTCQB.
| Period | Low Trading Price ($) | High
Trading Price ($) | ||||||
| Year Ending December 31, 2022 | ||||||||
| First Quarter (through March 31, 2022) | $ | 0.10 | $ | 0.11 | ||||
| Year Ending December 31, 2021 | ||||||||
| Fourth Quarter (December 31, 2021) | $ | 0.11 | $ | 0.12 | ||||
| Third Quarter (September 30, 2021) | $ | 0.14 | $ | 0.15 | ||||
| Second Quarter (June 30, 2021) | $ | 0.17 | $ | 0.19 | ||||
| First Quarter (March 31, 2021) | $ | 0.16 | $ | 0.18 | ||||
| Year Ending December 31, 2020 | ||||||||
| Fourth Quarter (December 31, 2020) | $ | 0.06 | $ | 0.09 | ||||
| Third Quarter (September 30, 2020) | $ | 0.11 | $ | 0.11 | ||||
| Second Quarter (June 30, 2020) | $ | 0.16 | $ | 0.16 | ||||
| First Quarter (March 31, 2020) | $ | 0.09 | $ | 0.09 | ||||
114
As of March 25, 2022, there were 123 holders of record of our Common Shares.
There are no restrictions in the Company’s articles, by-laws or elsewhere, which would prevent the Company from paying dividends. No dividends have been declared or paid on the Common Shares in the last five fiscal years, and it is not expected that dividends will be declared or paid in the immediate or foreseeable future. Consequently, to date there have been no distributions made by the Company. The policy of the Board is to reinvest all available funds in operations. The Board will reassess this policy from time to time. Any decision to pay dividends on the Common Shares will be made by the Board based on the assessment of, among other factors, earnings, capital requirements and the operating and financial condition of Company.
The following table sets forth securities authorized for issuance under the Stock Option Plan and the DSU Plan as of December 31, 2021. Figures below are presented on an as-converted basis.
(1) The total number of Common Shares that may be reserved and available for issuance under the Stock Option Plan shall not exceed the number of Common Shares equal to twenty percent (20%) of the total issued and outstanding Common Shares from time to time less any Common Shares reserved for issuance under the DSU Plan.
(2) The weighted-average exercise price reported herein does not take into account the DSUs awarded under the DSU Plan.
ITEM 10. RECENT SALES OF UNREGISTERED SECURITIES
The following information represents securities sold by the Company within the past three years through April 4, 2022 which were not registered under the Securities Act. Included are new issues, securities issued in exchange for property, services or other securities and new securities resulting from the modification of outstanding securities. The Company sold all of the securities listed below pursuant to the exemption from registration provided by Section 4(a)(2) of the Securities Act, or Regulation D or Regulation S promulgated thereunder.
2019
On January 2, 2019, the Company granted 1,000,000 Stock Options with an exercise price per Stock Option of C$0.25 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On January 22, 2019, the Company completed a private placement of 9,560,000 units at C$0.23 per unit, for gross proceeds of C$2,198,800. Each unit consisted of one Common Share and one Common Share purchase warrant. Each warrant entitles the holder to purchase one Common Share at an exercise price of C$0.48 each at any time for five years following closing of the private placement, subject to earlier expiry upon 30 days’ notice if, at any time after four months from closing (until January 23, 2024), the 20-day VWAP of Common Shares is greater than C$1.00, and ProMIS may accelerate the expiry of the Warrants by issuing a press release announcing the reduced term and expiry not less than 30 days after the publication date of such press release. In connection with the offering, the Company also issued a total of 164,500 finder’s warrants equal to 7% of the number of units sold to purchasers introduced by such finders. The finder’s warrants have the same term as the offering warrants.
115
On March 15, 2019, the Company issued 250,000 Common Shares at a price per share of C$0.30 for total aggregate consideration of C$75,000 upon the exercise of warrants.
On March 26, 2019, the Company issued 87,500 Common Shares at a price per share of C$0.20 for total aggregate consideration of C$17,500 upon the exercise of warrants.
In June 2019, the Company closed a private placement, consisting of 4,680,000 Units at C$0.25 for gross proceeds of C$1,170,000. Each Unit consisted of one Common Share and one warrant, with each warrant entitling the holder thereof to purchase one Common Share at an exercise price of C$0.35 per Common Share at any time for five years (until June 26, 2024) following the closing of the private placement.
On September 19, 2019, the Company granted 250,000 Stock Options with a price per Stock Option of C$0.23 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On December 31, 2019, the Company completed a private placement of 14,001,664 Units at C$0.20 per Common Share for total gross proceeds of C$2,800,333, issued in two tranches completed on November 15 and December 31, 2019. Each Unit consisted of one Common Share and one warrant, with each warrant entitling the holder thereof to purchase one Common Share at an exercise price of C$0.35 per Common Share at any time up to five years following closing of the private placement. In connection with the offering, the Company also issued a total of 162,400 finder’s warrants equal to 7% of the number of units sold to purchasers introduced by such finders. The finder’s warrants have the same term as the offering warrants.
2020
On February 25, 2020, the Company reported a total of 4,790,251 of the Common Share purchase warrants issued on February 10 and February 21, 2017 in a non-brokered private placement were exercised at a price of C$0.20 per Common Share for gross proceeds of C$958,051, which warrants expired on February 21, 2020.
On February 28, 2020, the Company granted 400,000 Stock Options with an exercise price per Stock Option of C$0.20 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On March 24, 2020, the Company announced that it had received TSX approval to reprice to C$0.13 each, in aggregate, 44,182,530 outstanding warrants issued between August 9, 2017 and December 30, 2019, all expiring 5 years from the original date of issuance. The warrant repricing period was effective from April 8, 2020 to May 22, 2020. In April and May 2020, the Company received gross proceed of C$1,239,195 from the exercise of 9,532,276 warrants from the warrant repricing program. At the end of the warrant repricing period, the warrants reverted to the original exercise price. All other terms of the warrants remain unchanged.
On April 15, 2020, the Company granted 150,000 Stock Options with an exercise price per Stock Option of C$0.15 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On October 23, 2020, the Company proceeded with a private placement offering (the “SW Offering”) of 16,776,781 special warrants (“Special Warrant”) at a price of C$0.12 per Special Warrant, for aggregate gross proceeds of up to C$3,000,000. The SW Offering closed in two tranches: the first in the amount of 13,819,581 Special Warrants closed on November 5, 2020 raising gross proceeds of $1,658,349.72; and the second in the amount of 2,400,000 Special Warrants closed on November 16, 2020, raising gross proceeds of $288,000 (the November 5 and 16, 2020 closings collectively, the “Closing”). Each Special Warrant is exercisable without payment of any additional consideration by the holder, into one unit (“Special Unit”), with each Special Unit consisting of one Common Share and one transferable Common Share purchase warrant (“Unit Warrant”). Each Unit Warrant entitles the holder thereof to acquire one Common Share (“Unit Warrant Share”) at an exercise price of C$0.20 per Unit Warrant Share for a period of 60 months after the Closing, subject to acceleration of the expiry date described as follows. If at any time after the expiry of the four-month hold period applicable to the Unit Warrants, the twenty-day VWAP of the Common Shares on the TSX, or such other exchange on which the Common Shares may be listed, is greater than C$0.60, the Company may deliver a notice to the holders of Unit Warrants accelerating the expiry date to a date that is not less than 30 days following the date of such notice. In connection with Offering, the Company issued 487,200 compensation warrants (“Compensation Warrants”) on November 5, 2020 and an additional 70,000 Compensation Warrants on November 16, 2020 for a total of 557,000 Compensation Warrants. The Compensation Warrants have the same terms as the Unit Warrants, except that the compensation warrants were immediately issued on November 5 and 16, 2020.
The Special Warrants will be deemed to be automatically exercised at 1:00 p.m. PT (the “Deemed Exercise Time”) on the earlier of the date (the “Conversion Date”) that is (i) the third business day after a receipt for a final prospectus qualifying the distribution of the shares and Unit Warrants issuable upon the conversion of the Special Warrants and (ii) 4 months and one day after the issue date of the Special Warrants. The Special Warrants will be deemed to have been exercised, delivered and surrendered by the holder thereof immediately prior to the Deemed Exercise Time without any further action on the part of the holder. The Company filed a preliminary short form prospectus dated November 26, 2020 to qualify the distribution of certain Units Warrants upon deemed conversion of the Special Warrants. However, the Special Warrants automatically converted on March 5, 2021 and March 17, 2021, as applicable, during the review process. The Corporation therefore did not file a final short form prospectus and a final receipt was not issued.
116
2021
On March 5 and 17, 2021, the Special Warrants issued by the Company in the SW Offering automatically converted, without payment of any additional consideration by the holder of the Special Warrants, into Special Units of the Company consisting of one Common Share and one Unit Warrant, pursuant to the terms of the Special Warrant. Upon the conversion of the Special Warrants, the holders of the Special Warrants were issued a total of 16,776,781 Common Shares and 16,776,781 Unit Warrants.
On March, 22, 2021, the Company completed a private placement for gross proceeds of $7 million by debentures convertible into Common Shares, at the option of the debenture holder, at $0.10 per Common Share and accrue interest at 1% per annum, which is payable annually. Interest may be paid in cash or in Common Shares, at the option of ProMIS (such number of Common Shares area to be determined by dividing the interest due by the 5-day VWAP of the Common Shares). The Debentures mature on March 22, 2026, and ProMIS has the option, prior to the maturity date, to force conversion of the Debentures at the conversion price upon raising $50 million in equity and/or debt cumulatively. At maturity ProMIS may redeem the outstanding principal amount of the Debentures in either cash or Common Shares at the then current 5-day VWAP less a 10% discount, or at its election, a combination thereof.
On March 30, 2021, the Company granted 1,500,000 Stock Options with an exercise price per Stock Option of C$0.17 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On May 14, 2021, the Company granted 750,000 Stock Options with an exercise price per Stock Option of C$0.18 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On June 30, 2021, the Company granted 50,000 Stock Options with an exercise price per Stock Option of C$0.21 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On June 30, 2021, the Company granted 2,437,500 Stock Options with an exercise price per Stock Option of C$0.30 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On August 12, 2021, the Company granted 100,000 Stock Options with an exercise price per Stock Option of C$0.24 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On August 25, 2021, the Company completed a public offering of 125,781,250 units at a price of $0.16 for gross proceeds of $20,125,000 and the issuance of 8,804,687 compensation warrants with a strike price of $0.16. Each unit consisted of one Common Share and one-quarter purchase warrant. Each purchase warrant entitles the holder thereof to purchase one common share at an exercise price of $0.21 per share at any time for five years.
On September 1, 2021, the Company granted 500,000 Stock Options with an exercise price per Stock Option of C$0.21 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On September 22, 2021, the Company granted 3,500,000 Stock Options with an exercise price per Stock Option of C$0.19 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On November 12, 2021, the Company granted 500,000 Stock Options with an exercise price per Stock Option of C$0.18 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
On December 9, 2021, the Company granted 2,500,000 Stock Options with an exercise price per Stock Option of C$0.14 to certain directors, executives and employees. No consideration was received by the Company for the issuance.
2022
On February 10, 2022, the Company granted 6,750,000 Stock Options with an exercise price per Stock Option of C$0.14 to certain directors, advisors, executives and employees. No consideration was received by the Company for this issuance.
On February 14, 2022, the Company granted 500,000 Stock Options with an exercise price per Stock Option of C$0.14 to certain employees. No consideration was received by the Company for this issuance.
117
ITEM 11. DESCRIPTION OF THE REGISTRANT’S SECURITIES TO BE REGISTERED
Description of the Company’s Securities
This Registration Statement relates to the Company’s Common Shares. The Company is authorized to issue an unlimited number of Common Shares, of which there are 431,731,592 Common Shares issued and outstanding as of March 25, 2022, and an unlimited number of preferred shares, of which there no preferred shares issued and outstanding as of March 25, 2022. The following description may not be complete and is subject to, and qualified in its entirety by reference to, the terms and provisions of our articles and by-laws, as amended (the “Constating Documents”), which are included as exhibits to this Registration Statement.
As of March 25, 2022, the Common Shares collectively represent 100% of our total issued and outstanding shares and 100% of the voting power attached to all of our issued and outstanding shares. The Company refers to its Common Shares as “shares.”
Common Shares
Voting Rights
The holders of Common Shares shall be entitled to receive notice of all meetings of shareholders, and to attend, vote and speak at such meetings, except those meetings at which only holders of another specified class or series of shares of the Company are entitled to vote separately as a class or series. A quorum for a meeting of Shareholders shall be two shareholders, or two proxyholders representing shareholders, or any combination thereof, holding not less than one-twentieth of the issued shares entitled to be voted at the meeting. On all matters upon which holders of shares are entitled to vote, each Common Share is entitled to one vote per Common Share. Unless a different majority is required by law or the Constating Documents, resolutions to be approved by holders of shares require approval by a simple majority of the total number of votes of all shares cast at a meeting of Shareholders at which a quorum is present.
Dividend Rights
There are no restrictions in the Company’s articles or elsewhere, which would prevent the Company from paying dividends. No dividends have been declared or paid on the Common Shares of the Company in the last five fiscal years, and it is not expected that dividends will be declared or paid in the immediate or foreseeable future. Consequently, to date there have been no distributions made by the Company. The policy of the Board is to reinvest all available funds in operations. The Board will reassess this policy from time to time. Any decision to pay dividends on the Common Shares of the Company will be made by the Board based on the assessment of, among other factors, earnings, capital requirements and the operating and financial condition of the Company.
Liquidation Rights
In the event of the liquidation, dissolution or winding-up of the Company or any other distribution of the Company’s assets for the purpose of winding up the Company’s affairs, after the payment of dividends declared but unpaid, the holders of Common Shares shall be entitled pari passu to receive any remaining property of the Company.
Pre-emptive and Redemption Rights
Holders of Common Shares do not have any pre-emptive or redemption rights.
Shareholder Rights Plan
On January 18, 2016 the Board adopted the Shareholder Rights Plan effective January 22, 2016. The Shareholder Rights Plan had an initial three-year term to the end of ProMIS’ 2019 annual shareholders meeting. At the Corporation’s annual shareholders meeting held June 27, 2019, the shareholders ratified and approved the Shareholder Rights Plan, as amended and extended, for continuation for a further three-year period – specifically until the end of the annual shareholders meeting to be held in June 2022. A copy of the Shareholder Rights Plan, as amended and extended, is filed under the Corporation’s profile at www.sedar.com.
The Shareholder Rights Plan is intended to ensure, to the extent possible, that the ProMIS’ Board and shareholders have adequate time to consider and evaluate any unsolicited takeover bid and to identify, solicit, develop and negotiate any value enhancing alternatives that would be considered appropriate. This will encourage fair treatment of ProMIS’ shareholders in connection with any unsolicited takeover bid. The Shareholder Rights Plan was not adopted in response to, or in anticipation of, any acquisition or take-over offer and is not intended to prevent a take-over of ProMIS, to secure continuity of current management or the directors in office or to deter fair offers for the Common Shares of ProMIS.
118
The Board authorized the issuance of one right in respect of each Common Share of ProMIS outstanding at 5:00 p.m. (ET) on January 22, 2016 and each Common Share issued thereafter. The rights will become exercisable if a person, together with their affiliates, associates and joint actors, acquires or announces an intention to acquire beneficial ownership of Common Shares which, when aggregated with its holdings, total 20% or more of the outstanding Common Shares of ProMIS (determined in the manner set out in the Shareholder Rights Plan). Following the acquisition of 20% or more of the outstanding Common Shares, each right held by a person other than the acquiring person and its affiliates, associates and joint actors would, upon exercise, entitle the holder to purchase that number of Common Shares at a substantial discount to the market price of the Common Shares at that time.
The Shareholder Rights Plan permits the acquisition of control of ProMIS through a “permitted bid”, a “competing permitted bid” or a negotiated transaction. A “permitted bid” is one that, among other things, is made to all holders of Common Shares, is open for a minimum of 120 days and is subject to an irrevocable minimum tender condition of at least 50% of the Common Shares held by independent shareholders. The Board has the discretion to defer the time at which the rights become exercisable and to waive the application of the Shareholder Rights Plan.
Preferred Shares
The preferred shares of ProMIS may be issued in one or more series and the directors are authorized to fix the number of preferred shares in each series and to determine the designation, rights, privileges, restrictions and conditions attached to the preferred shares of each series. ProMIS preferred shares rank on parity with the Common Shares with respect to the payment of dividends unless one or more series of ProMIS preferred shares are entitled to cumulative dividends. ProMIS preferred shares also rank on parity with the preferred shares of every other series and are entitled to a priority over any other class of shares ranking junior to the ProMIS preferred shares, and all series of preferred shares participate rateably with respect to the distribution of assets upon the liquidation, dissolution or winding-up of ProMIS.
ITEM 12. INDEMNIFICATION OF DIRECTORS AND OFFICERS
Indemnification of our officers and our directors is subject to the provisions of the CBCA, principally section 124 of the CBCA, which provides as follows:
Indemnification
(1) A corporation may indemnify a director or officer of the corporation, a former director or officer of the corporation or another individual who acts or acted at the corporation’s request as a director or officer, or an individual acting in a similar capacity, of another entity, against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with the corporation or other entity
Advance of Costs
(2) A corporation may advance moneys to a director, officer or other individual for the costs, charges and expenses of a proceeding referred to above in subsection (1). The individual shall repay the moneys if the individual does not fulfil the conditions of subsection (3).
Limitation
(3) A corporation may not indemnify an individual under subsection (1) unless the individual:
(a) acted honestly and in good faith with a view to the best interests of the corporation, or, as the case may be, to the best interests of the other entity for which the individual acted as director or officer or in a similar capacity at the corporation’s request; and
(b) in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the individual had reasonable grounds for believing that the individual’s conduct was lawful.
119
Indemnification in Derivative Actions
(4) A corporation may with the approval of a court, indemnify an individual referred to in subsection (1), or advance moneys under subsection (2), in respect of an action by or on behalf of the corporation or other entity to procure a judgment in its favor, to which the individual is made a party because of the individual’s association with the corporation or other entity as described in subsection (1) against all costs, charges and expenses reasonably incurred by the individual in connection with such action, if the individual fulfils the conditions set out in subsection (3).
Right to Indemnity
(5) Despite subsection (1), an individual referred to in that subsection is entitled to indemnity from the corporation in respect of all costs, charges and expenses reasonably incurred by the individual in connection with the defense of any civil, criminal, administrative, investigative or other proceeding to which the individual is subject because of the individual’s association with the corporation or other entity as described in subsection (1), if the individual seeking indemnity:
(a) was not judged by the court or other competent authority to have committed any fault or omitted to do anything that the individual ought to have done; and
(b) fulfils the conditions set out in subsection (3).
Insurance
(6) A corporation may purchase and maintain insurance for the benefit of an individual referred to in subsection (1) against any liability incurred by the individual:
(a) in the individual’s capacity as a director or officer of the corporation; or
(b) in the individual’s capacity as a director or officer, or similar capacity, of another entity, if the individual acts or acted in that capacity at the corporation’s request.
Application to Court
(7) A corporation, an individual or an entity referred to in subsection (1) may apply to a court for an order approving an indemnity under this section and the court may so order and make any further order that it sees fit.
Notice to Director
(8) An applicant under subsection (7) shall give the Director notice of the application and the Director is entitled to appear and be heard in person or by counsel.
120
Other Notice
(9) On an application under subsection (7) the court may order notice to be given to any interested person and the person is entitled to appear and be heard in person or by counsel.
Our by-laws provide subject to the CBCA, we may indemnify a director or officer or a former director or officer or a corporation of which we are or were or shareholder or creditor and their heirs and legal representatives of such person against all costs, charges, and expenses including and amount to be paid to settle an action or satisfy a judgment, reasonably incurred in respect of any civil, criminal or administrative action or proceeding to which they are made a party by reason of being or having been a director or officer of us or a director or officer of any such corporation. Each director and officer of on being elected and appointed shall be deemed to have contracted with us on the terms of this indemnity. The failure of a director or officer to comply with the provisions of the CBCA, the articles or the by-laws shall not invalidate any indemnity to which they are entitled under our by-laws.
We have entered into indemnification agreements or employment agreements containing indemnification provisions with certain of our executive officers and our directors. See “Consulting and Employment Agreements” above for more information about our employment agreements. Under these indemnification provisions, an indemnified person is entitled, subject to the terms and conditions thereof, to the right of indemnification by the Company for certain expenses to the fullest extent permitted by applicable law. We believe that these indemnification agreements are necessary to attract and retain qualified individuals to serve as directors and executive officers.
The directors may cause us to purchase and maintain insurance for the benefit of any person who is or was serving as a director, officer, employee or agent of us or as a director, officer, employee or agent of any corporation of which we are or were a shareholder and their heirs or personal representatives against any liability incurred as a director, officer, employee or agent.
We have an insurance policy covering our directors and officers, within the limits and subject to the limitations of the policy, with respect to certain liabilities arising out of claims based on acts or omissions in their capacities as directors or officers.
ITEM 13. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required to be included in this Registration Statement are included in Item 15 and begin on page F-1.
ITEM 14. CHANGES IN AND DISAGREEMENTS WITH AUDITORS ON ACCOUNTING AND FINANCIAL DISCLOSURE
On December 10, 2021, the Company engaged Baker Tilly US, LLP (“Baker Tilly”) to perform an audit (the “U.S. Audit”) in accordance with the standards of the Public Company Accounting Oversight Board of the Company’s financial statements prepared in conformity with accounting principles generally accepted in the U.S. for its fiscal years ended December 31, 2020 and 2021, which financial statements are included in this Registration Statement. Baker Tilly’s engagement was recommended to the Company’s Board by the Audit Committee and approved by the Board.
Prior to engaging Baker Tilly to conduct the U.S. Audit, the Company had not consulted Baker Tilly regarding the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements, nor did the Company consult with Baker Tilly regarding any matter that was either the subject of a disagreement (as defined in paragraph (a)(1)(iv) of Item 304 of Regulation S-K) or a reportable event (as described in paragraph (a)(1)(v) of Item 304 of Regulation S-K).
As of the date of filing of this Registration Statement, PricewaterhouseCoopers LLP remains engaged as the Company’s independent auditor in Canada. PricewaterhouseCoopers LLP has served as the Company’s auditors since 2004 and has conducted its most recent audits under Canadian Auditing Standards of the Company’s financial statements prepared in accordance with International Financial Reporting Standards. In connection with the effectiveness of this Registration Statement, if and when such effectiveness may occur, the Company anticipates that it will request PricewaterhouseCoopers LLP to resign as its auditor.
121
ITEM 15. FINANCIAL STATEMENTS AND EXHIBITS
ProMIS Neurosciences Inc.
Years Ended December 31, 2021 and 2020
Index to Consolidated Financial Statements
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of ProMIS Neurosciences Inc. and Subsidiary
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ProMIS Neurosciences Inc. and Subsidiary (the “Company”) as of December 31, 2021 and 2020, and the related consolidated statements of operations and comprehensive loss, changes in shareholders’ equity (deficit), and cash flows for each of the two years in the period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
|
/s/ Baker Tilly US, LLP
We have served as the Company’s auditor since 2021. |
|
|
Tewksbury, Massachusetts | |
| April 4, 2022 | |
F-2
ProMIS Neurosciences Inc.
(expressed in US dollars, except share and per share amounts)
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash | $ | 16,943,905 | $ | 806,887 | ||||
| Short-term investments | 33,248 | 32,963 | ||||||
| Prepaid expenses and other current assets | 737,316 | 133,022 | ||||||
| Total current assets | 17,714,469 | 972,872 | ||||||
| Property and equipment, net | 4,671 | 78,111 | ||||||
| Intangible assets, net | 27,614 | 32,637 | ||||||
| Other assets | — | 2,353 | ||||||
| Total assets | $ | 17,746,754 | $ | 1,085,973 | ||||
| Liabilities and Shareholders' Equity (Deficit) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 408,981 | $ | 437,441 | ||||
| Accrued liabilities | 520,093 | 46,201 | ||||||
| Deferred compensation | — | 1,398,989 | ||||||
| Total current liabilities | 929,074 | 1,882,631 | ||||||
| Convertible debt, net of issuance costs and debt discount | 3,906,057 | — | ||||||
| Derivative liability | 5,379,878 | — | ||||||
| Warrant liability | 1,871,687 | — | ||||||
| Total liabilities | 12,086,696 | 1,882,631 | ||||||
| Commitments and contingencies (Note 16) | ||||||||
| Shareholders' equity (deficit): | ||||||||
| Common shares, no par value, unlimited shares authorized, 431,731,591 and 289,730,760 shares issued and outstanding as of December 31, 2021 and 2020, respectively | — | — | ||||||
| Additional paid-in capital | 68,039,178 | 51,655,168 | ||||||
| Accumulated other comprehensive loss | (187,919 | ) | (50,731 | ) | ||||
| Accumulated deficit | (62,191,201 | ) | (52,401,095 | ) | ||||
| Total shareholders' equity (deficit) | 5,660,058 | (796,658 | ) | |||||
| Total liabilities and shareholders' equity (deficit) | $ | 17,746,754 | $ | 1,085,973 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
ProMIS Neurosciences Inc.
Consolidated Statements of Operations and Comprehensive Loss
(expressed in US dollars, except share and per share amounts)
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 4,627,386 | $ | 2,224,650 | ||||
| General and administrative | 3,663,707 | 2,026,957 | ||||||
| Total operating expenses | 8,291,093 | 4,251,607 | ||||||
| Loss from operations | (8,291,093 | ) | (4,251,607 | ) | ||||
| Change in fair value of financial instruments | (1,095,636 | ) | — | |||||
| Interest expense on convertible debt | (416,286 | ) | — | |||||
| Other income | 12,909 | 1,327 | ||||||
| Net loss | (9,790,106 | ) | (4,250,280 | ) | ||||
| Other comprehensive loss | ||||||||
| Foreign currency translation adjustment | (137,188 | ) | (72,803 | ) | ||||
| Comprehensive loss | $ | (9,927,294 | ) | $ | (4,323,083 | ) | ||
| Net loss per share, basic and diluted | $ | (0.03 | ) | $ | (0.01 | ) | ||
| Weighted-average shares outstanding of common shares, basic and diluted | 347,137,045 | 285,599,827 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
ProMIS Neurosciences Inc.
Consolidated Statements of Changes in Shareholders’ Equity (Deficit)
(expressed in US dollars, except share amounts)
| Common Shares | ||||||||||||||||||||||||
| Shares | Amount | Additional Paid-in Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Total | |||||||||||||||||||
| Balance, January 1, 2020 | 275,408,233 | $ | — | $ | 48,435,848 | $ | 22,072 | $ | (48,150,815 | ) | $ | 307,105 | ||||||||||||
| Exercise of warrants | 14,322,527 | — | 1,608,090 | — | — | 1,608,090 | ||||||||||||||||||
| Issuance of special warrants - net of issuance costs of $226,703 | — | — | 1,256,828 | — | — | 1,256,828 | ||||||||||||||||||
| Share-based compensation | — | — | 354,402 | — | — | 354,402 | ||||||||||||||||||
| Foreign currency translation | — | — | — | (72,803 | ) | — | (72,803 | ) | ||||||||||||||||
| Net loss | — | — | — | — | (4,250,280 | ) | (4,250,280 | ) | ||||||||||||||||
| Balance, December 31, 2020 | 289,730,760 | — | 51,655,168 | (50,731 | ) | (52,401,095 | ) | (796,658 | ) | |||||||||||||||
| Conversion of special warrants | 16,219,581 | — | — | — | — | — | ||||||||||||||||||
| Issuance of common shares, net of issuance costs of $1,665,099 | 125,781,250 | — | 15,868,381 | — | — | 15,868,381 | ||||||||||||||||||
| Share-based compensation | — | — | 515,629 | — | — | 515,629 | ||||||||||||||||||
| Foreign currency translation | — | — | — | (137,188 | ) | — | (137,188 | ) | ||||||||||||||||
| Net loss | — | — | — | — | (9,790,106 | ) | (9,790,106 | ) | ||||||||||||||||
| Balance, December 31, 2021 | 431,731,591 | $ | — | $ | 68,039,178 | $ | (187,919 | ) | $ | (62,191,201 | ) | $ | 5,660,058 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
ProMIS Neurosciences Inc.
Consolidated Statements of Cash Flows
(expressed in US dollars)
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (9,790,106 | ) | $ | (4,250,280 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Share-based compensation | 515,629 | 354,402 | ||||||
| Foreign currency exchange loss | 85,066 | — | ||||||
| Change in fair value of derivative liability | 1,936,191 | — | ||||||
| Change in fair value of warrant liability | (840,555 | ) | — | |||||
| Depreciation of property and equipment | 40,576 | 6,726 | ||||||
| Gain on sale of property and equipment | (59,157 | ) | — | |||||
| Amortization of debt discount and issuance costs | 366,018 | — | ||||||
| Amortization of intangible assets | 5,249 | 4,955 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (600,635 | ) | 113,017 | |||||
| Accounts payable | (31,654 | ) | 68,933 | |||||
| Accrued liabilities | 471,463 | (115,978 | ) | |||||
| Deferred compensation | (1,403,468 | ) | 585,693 | |||||
| Net cash used in operating activities | (9,305,383 | ) | (3,232,532 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of short-term investments | (33,102 | ) | (31,220 | ) | ||||
| Maturity of short-term investment | 33,069 | 31,064 | ||||||
| Proceeds from sale of property and equipment | 98,335 | — | ||||||
| Purchase of property and equipment | (6,044 | ) | (80,705 | ) | ||||
| Other investing activities | 2,360 | (2,228 | ) | |||||
| Net cash provided by (used in) investing activities | 94,618 | (83,089 | ) | |||||
| Cash flows from financing activities | ||||||||
| Proceeds from convertible debt | 6,915,199 | — | ||||||
| Proceeds from issuance of common share units, net of issuance costs | 15,868,381 | — | ||||||
| Proceeds from issuance of warrants | 2,739,221 | — | ||||||
| Proceeds from issuance of common shares from exercise of warrants | — | 1,608,090 | ||||||
| Proceeds from issuance of Special Warrants - net of issuance costs | — | 1,256,828 | ||||||
| Net cash provided by financing activities | 25,522,801 | 2,864,918 | ||||||
| Effect of exchange rates on cash | (175,018 | ) | (24,235 | ) | ||||
| Net increase (decrease) in cash | 16,137,018 | (474,938 | ) | |||||
| Cash at beginning of year | 806,887 | 1,281,825 | ||||||
| Cash at end of year | $ | 16,943,905 | $ | 806,887 | ||||
| Noncash financing activities | ||||||||
| Issuance of compensation warrants in consideration of issuance costs | $ | 957,947 | $ | 29,618 | ||||
| Fair value adjustment for modification of warrants | $ | — | $ | 85,005 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
ProMIS Neurosciences Inc.
Notes to Consolidated Financial Statements
(expressed in US dollars, except share and per share amounts)
| 1. | DESCRIPTION OF BUSINESS |
Business Description
ProMIS Neurosciences Inc. (the “Company” or “ProMIS”) is applying its patented technology platform to build a portfolio of antibody therapies, therapeutic vaccines, and other antibody-based therapies in neurodegenerative diseases and other mis-folded protein diseases, including Alzheimer’s disease multiple system atrophy, and amyotrophic lateral sclerosis. The Company also plans to investigate additional synucleinopathies, including Parkinson’s disease and dementia with Lewy bodies, frontotemporal lobar degeneration, progressive supranuclear palsy, corticobasal degeneration and schizophrenia. These diseases share a common biologic cause – misfolded versions of proteins that otherwise perform a normal function, become toxic and kill neurons, resulting in disease. ProMIS’ technology platform is an example of the advances in drug discovery enabled by computational power, in silico discovery, and/or artificial intelligence. ProMIS believes this platform provides a potential advantage by selectively targeting the toxic misfolded proteins with therapeutics or detecting them with diagnostics.
The Company was incorporated on January 23, 2004 under the Canada Business Corporations Act and is located at 1920 Yonge Street, Toronto, Ontario. The Company’s common shares are traded on the Toronto Stock Exchange (“TSX”) under the symbol PMN and on the OTCQB Venture Market under the symbol ARFXF. The Company has a wholly-owned U.S. subsidiary, ProMIS Neurosciences (US) Inc. (“ProMIS USA”), which was incorporated in January 2016 in the State of Delaware. As of December 31, 2021, ProMIS USA has had no activity and has no financial impact on the Company’s consolidated financial statements.
The success of the Company is dependent on obtaining the necessary regulatory approvals of its product candidates, marketing its products and achieving profitable operations. The continuation of the research and development activities and the commercialization of its products, if approved, are dependent on the Company’s ability to successfully complete these activities and to obtain additional financing through a combination of financing activities and operations. It is not possible to predict either the outcome of future research and development or commercialization programs, or the Company’s ability to fund these programs.
COVID-19
Impacts resulting from the COVID-19 pandemic have resulted in a widespread health crisis that has already adversely affected the economies and financial markets of many countries around the world. The international response to the spread of COVID-19 has led to significant restrictions on travel; temporary business closures; quarantines; global stock market and financial market volatility; a general reduction in consumer activity; operating, supply chain and project development delays and disruptions; and declining trade and market sentiment; all of which have and could further affect the world economy.
The extent to which the novel coronavirus may impact the Company’s business, preclinical research and development activities will depend on future developments which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, travel restrictions and social distancing in Canada, the United States and other countries, business closures or business disruptions and the effectiveness of actions taken by governments around the globe to contain and treat the disease. International scientific conferences at which the Company has been invited to present have been postponed, cancelled or will be held online instead, which diminishes exposure and the opportunity to meet with collaborators and potential partners. These scientific conferences have started to be held in person with an option to attend online. Vendors performing work for the Company have remained open, although they have indicated that their timelines are now somewhat longer. The current global uncertainty and its effect on the local and global economies may also have an adverse effect on the Company’s ability to secure additional financing to continue its research and development programs.
F-7
Liquidity Risk
The accompanying consolidated financial statements were prepared on a going concern basis, which assumes that the Company will continue its operations for the foreseeable future and will be able to realize its assets and discharge its liabilities in the normal course of business. To date, the Company has not generated revenues from its activities. The Company had a net loss of $9,790,106, for the year ended December 31, 2021 and an accumulated deficit of $62,191,201 as of December 31, 2021. In March 2021, the Company completed a $7.0 million private placement of convertible unsecured debentures (Note 10). In August 2021, the Company completed a public offering on the TSX, with gross proceeds of $20,125,000 (Note 11). Available funds, including the proceeds from the March 2021 financing and August 2021 financing, are expected to be sufficient to fund the Company’s operating expenses for at least 12 months from the date the consolidated financial statements are issued. However, additional funding will be necessary to fund future research, pre-clinical and clinical activities. The Company will seek additional funding through public financings, debt financings, collaboration agreements, strategic alliances and licensing agreements. Although the Company has been successful in raising capital in the past, there is no assurance of success in obtaining such additional financing on terms acceptable to us, if at all, and there is no assurance that the Company will be able to enter into collaborations or other arrangements. If the Company is unable to obtain funding, it could force delays, reduce or eliminate research and development programs, product portfolio expansion or commercialization efforts, which could adversely affect future business prospects, and the ability to continue operations.
The Company may continue to incur net losses for at least the next several years as the Company advances its product candidates. The Company is actively pursuing additional financing to further develop certain of the Company’s scientific initiatives, but there is no assurance these initiatives will be successful, timely or sufficient.
| 2. | BASIS OF PRESENTATION AND Summary of Significant Accounting Policies |
Basis of Presentation
The accompanying consolidated financial statements have been prepared in conformity with generally accepted accounting principles in the United States of America (“GAAP”). Any reference in these notes to applicable guidance is meant to refer to the authoritative GAAP as found in the Accounting Standards Codification (“ASC”) and as amended by Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (“FASB”).
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make certain estimates, judgements and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates and assumptions made in the accompanying consolidated financial statements include, but are not limited to, the valuation of intangible assets, accrual for research and development expenses, the valuation of share-based compensation and the valuation of warrants, and the valuation of warrant liabilities and embedded derivative liabilities. Actual results could differ from those estimates, and such differences could be material to the consolidated financial statements.
F-8
Segment Information
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker (“CODM”), or decision-making group, in making decisions on how to allocate resources and assess performance. The Company has one operating segment and its Chief Executive Officer and Executive Chairman of the Board of Directors collectively serve as the CODM. Substantially all of the Company’s assets are located in Canada.
Foreign Currency
Comprehensive loss is defined as a change in equity of a business enterprise during a period, resulting from transactions from non-owner sources. The reporting currency of the Company is the United States dollar (“US$”) and the functional currency of the Company is the Canadian dollar (“C$”). The assets and liabilities of the Company are translated to US$ at exchange rates in effect at the balance sheet date. All income statement accounts are translated at average exchange rates. Resulting foreign currency translation adjustments are recorded directly in accumulated other comprehensive income (loss) as a separate component of shareholders’ equity (deficit). Transaction gains and losses that arise from exchange rate fluctuations on transactions denominated in a currency other than the functional currency are included in general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss when realized and are not material for the years ended December 31, 2021 and 2020.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less at the date of purchase to be cash equivalents. As of December 31, 2021 and 2020 the Company had no cash equivalents.
Short-term Investments
Short-term investments consist of guaranteed certificates of deposit with a maturity greater than 90 days and up to one year at the time of purchase. Accordingly, all short-term investments are classified as current assets in the accompanying consolidated balance sheets. The short-term investment is being held as collateral for the Company’s credit cards.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk consist of cash and short-term investments. Cash is deposited in checking and money market accounts at accredited financial institutions, which at times, may exceed federally insured limits. The short-term investment is deposited in a guaranteed certificate of deposit with an accredited financial institution that guarantees 100% of the original amount invested. Management believes that these financial institutions are financially sound, and, accordingly, minimal credit risk exists with respect to these high-quality financial institutions. As of December 31, 2021, the Company has not experienced any losses on its cash or short-term investments.
F-9
Fair Value Measurements
FASB ASC 820, Fair Value Measurements and Disclosures, (“ASC 820”) defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, as established by ASC 820, of which the first two are considered observable and the last is considered unobservable:
| · | Level 1 – Observable inputs, such as unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date. |
| · | Level 2 – Inputs (other than Level 1 quoted prices) are either directly or indirectly observable inputs for similar assets or liabilities. These include quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities. |
| · | Level 3 – Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. Financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
The Company’s warrant liability and embedded derivative liability were classified as Level 3 financial instruments for the year ended December 31, 2021.
The carrying amounts of prepaid and other current assets, short-term investments, accounts payable, and accrued expenses are generally considered to be representative of their fair value based on the short-term nature of these financial instruments.
Impairment of Long-Lived Assets
The Company evaluates its long-lived assets, which consist of property and equipment and definite-lived intangible assets, for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds the fair value of the asset. No impairments have been identified as of December 31, 2021 and 2020.
F-10
Property and Equipment
Property and equipment, net are stated at cost less accumulated depreciation. Depreciation expense is recognized using the straight-line method over the estimated useful life of each asset. Laboratory and equipment are depreciated over two to five years. Computer equipment is depreciated over two to three years. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accompanying consolidated balance sheets and any resulting gain or loss is included in loss from operations in the accompanying consolidated statements of operations and comprehensive loss. Expenditures for repairs and maintenance are expensed as incurred.
Intangible Assets
Definite-lived intangible assets are stated at cost less accumulated amortization and any accumulated impairment losses. An intangible asset’s carrying amount is assessed for impairment whenever there is an indication that the asset may be impaired. The Company’s definite-lived intangible assets consist of acquired rights and patents. Intangible assets are amortized on a straight-line basis over the lesser of the life of the intangible asset or its estimated useful life, which is 15 years.
Derivative Liability
The Company evaluates its convertible debt, warrants or other contracts to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for in accordance with FASB ASC Topic 815, Derivatives and Hedging and Topic 480, Distinguishing Liabilities from Equity. The result of this accounting treatment is that the fair value of the embedded derivative, if required to be bifurcated, is marked-to-market at each balance sheet date and recorded as a liability. The change in fair value is recorded in the accompanying consolidated statements of operations and comprehensive loss as a component of other income or expense. Upon conversion or exercise of a derivative instrument, the instrument is marked to fair value at the conversion date and then that fair value is reclassified to equity.
Collaboration Arrangements
The Company may enter into collaboration arrangements with pharmaceutical and biotechnology partners. The Company analyzes its collaboration arrangements to assess whether they are within the scope of FASB ASC 808, Collaborative Arrangements, (“ASC 808”), to determine whether such arrangements involve joint operating activities performed by the parties that are both active participants in the activities and exposed to significant risks and rewards dependent on the commercial success of such activities. This assessment is performed throughout the life of the arrangement based on changes in responsibilities of all parties in the arrangement. ASC 808 does not provide guidance on the recognition of consideration exchanged or accounting for the obligations that may arise between parties. The Company concluded that ASC Topic 730, Research and Development, should be applied by analogy to payments between parties during the development activities of its collaboration arrangements.
General and Administrative
General and administrative expenses consist primarily of personnel costs including salary, bonus, employee-benefits and share-based compensation, costs incurred in development and protection of intellectual property, professional service fees, and other general overhead and facility costs, including rent, depreciation and amortization, which relate to the Company’s general and administrative functions.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred in connection with the development and research of the Company’s platform technology, as well as discovery program expenses. The Company expenses research and development costs as incurred. These expenses include:
| · | employee-related expenses, including salaries, related benefits and share-based compensation expense, for employees engaged in research and development functions; |
F-11
| · | external research and development expenses incurred under arrangements with third parties, such as contract research organizations (“CROs”), and consultants; |
| · | the cost of acquiring, developing, and manufacturing clinical study materials; and |
| · | Costs associated with preclinical and clinical activities and regulatory operations. |
Prepaid and Accrued Research and Development Expenses
Substantial portions of the Company’s pre-clinical trials are performed by third-party laboratories, medical centers, CROs and other vendors. These vendors generally bill monthly for services performed, or bill based upon milestone achievement. For preclinical studies, the Company accrues expenses based upon estimated percentage of work completed and the remaining contract milestones. At times, the Company is obligated to make upfront payments upon execution of research and development agreements. Upfront payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities are capitalized as prepaid expenses until such goods are delivered or the related services are performed. The Company estimates the period over which such services will be performed based on the terms of the agreements as well as the level of effort to be expended in each period. Sometimes the actual timing of performance or the level of effort varies from the estimate, and if that does occur, the Company will adjust the amounts recorded accordingly.
Patent Costs
All patent-related costs incurred in connection with filing and prosecuting patent applications are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss.
Warrants
The Company issues warrants on its common shares in connection with financings as well as for compensation of intermediaries and advisors. The Company accounts for warrants as either equity instruments or as liabilities depending on the specific terms of the warrant agreements in accordance with FASB ASC Topic 815, Derivatives and Hedging and Topic 480, Distinguishing Liabilities from Equity. When classified as equity, warrants are recorded within additional paid-in-capital. Warrants identified as meeting the definition of a derivative are recognized as a liability and treated in accordance with the derivative liability accounting policy described above.
Debt Issuance Costs
Debt issuance costs are specifically identifiable costs associated with issuance of a new debt instrument. Debt issuance costs are reported on the consolidated balance sheet as a direct deduction from the face amount of the related debt. Debt issuance costs are amortized to interest expense over the term of the related debt.
Share-based Compensation
Share-based compensation expense related to share awards granted to employees, directors and non-employees is recognized based on the grant-date estimated fair values of the awards using the Black- Scholes option pricing model (“Black-Scholes”). The value is recognized as expense ratably over the requisite service period, which is generally the vesting term of the award. The Company adjusts the expense for actual forfeitures as they occur. Share based compensation expense is classified in the accompanying consolidated statements of operations and comprehensive loss based on the function to which the related services are provided.
F-12
Black Scholes requires a number of assumptions, of which the most significant are expected volatility, expected option term (the time from the grant date until the options are exercised or expire) and risk-free rate. Expected volatility is determined using the historical volatility for the Company. The risk-free interest rate is based on the yield of Canadian government bonds with a remaining term equal to the expected life of the option. Expected dividend yield is zero because the Company has never paid cash dividends on common shares and the Company does not expect to pay cash dividends in the foreseeable future.
Income Taxes
The Company is a taxable entity under the Income Tax Act (Canada). Deferred income tax assets and liabilities are determined based on differences between the financial statement carrying amounts and the respective income tax bases of assets and liabilities, measured using substantively enacted income tax rates and laws that are expected to be in effect when the differences are expected to reverse. Deferred tax assets are recognized to the extent it is more likely than not that taxable income will be available against which the deferred tax asset can be utilized. Deferred tax assets are reduced by a valuation allowance if it is more likely than not that some portion or all of the deferred tax assets will not be realized.
The Company follows the provisions of ASC 740-10, Uncertainty in Income Taxes (“ASC 740-10”). The Company has not recognized a liability as a result of the implementation of ASC 740-10. A reconciliation of the beginning and ending amount of unrecognized tax benefits has not been provided since there is no unrecognized benefit since the date of adoption. The Company has not recognized interest expense or penalties as a result of the implementation of ASC 740-10. If there were an unrecognized tax benefit, the Company would recognize interest accrued related to unrecognized tax benefits in interest expense and penalties in operating expenses.
Basic and Diluted Net Loss Per Share
Basic net loss per common share is computed by dividing net loss by the weighted-average number of common shares outstanding during each period. Diluted net loss per share of common shares includes the effect, if any, from the potential exercise or conversion of securities, such as convertible debt, share options and warrants, which would result in the issuance of incremental shares of common shares. For diluted net loss per share, the weighted-average number of common shares is the same for basic net loss per share due to the fact that when a net loss exists, dilutive securities are not included in the calculation as the impact is anti-dilutive. For all periods presented, basic and diluted net loss per share are the same, as any additional share equivalents would be anti-dilutive. As the Company has reported a net loss for all periods presented, diluted net loss per common share is the same as basic net loss per common share.
F-13
Emerging Growth Company Status
The Company is an Emerging Growth Company, as defined in Section 2(a) of the Securities Act of 1933, as modified by the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”). Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act, until such time as those standards apply to private companies. The Company has elected to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date that it (i) is no longer an emerging growth company or (ii) affirmatively and irrevocably opts out of the extended transition period provided in the JOBS Act. As a result, these consolidated financial statements may not be comparable to companies that comply with the new or revised accounting pronouncements as of public company effective dates.
Recently Issued Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases (“Topic 842”), which requires lessees to recognize the following for all leases (with the exception of short-term leases) at the commencement date: a lease liability, which is a lessee’s obligation to make lease payments arising from a lease, measured on a discounted basis; and a right-of-use asset, which is an asset that represents the lessee’s right to use, or control the use of, a specified asset for the lease term. In July 2018, the FASB issued ASU 2018-11, Leases (“Topic 842”) Targeted Improvements, to amend certain aspects of Topic 842. These amendments provide entities with an additional (and optional) transition method to adopt Topic 842. Under this transition method, an entity initially applies the transition requirements in Topic 842 at that Topic’s effective date with the effects of initially applying Topic 842 recognized as a cumulative effect adjustment to the opening balance of retained earnings (or other components of equity or net assets, as appropriate) in the period of adoption. On April 8, 2020, the FASB changed the effective date of this standard applicable to the Company as an emerging growth company to January 1, 2022. The Company is currently evaluating the potential impact of adopting this standard on its consolidated financial statements.
In December 2019, the FASB issued ASU No 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (“Topic 740”), as part of its simplification initiative to reduce the cost and complexity in accounting for income taxes. The amendments in ASU 2019-12 removes certain exceptions related to the approach for intraperiod tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. ASU 2019-12 also amends other aspects of the guidance to help simplify and promote consistent application of GAAP. The guidance is effective for interim and annual periods beginning after December 15, 2020, with early adoption permitted. For emerging growth companies, the standard is effective for fiscal years beginning after December 15, 2021. The Company is currently evaluating the potential impact adopting ASU 2019-12 will have on the Company’s consolidated financial statements and related disclosures.
In August 2020, the FASB issued ASU No. 2020-06, Debt—Debt with Conversion and Other Options (“Subtopic 470-20 ”) and Derivatives and Hedging Contracts in Entity s Own Equity (“Subtopic 815-40 ”): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity. ASU 2020-06 will simplify the accounting for convertible instruments by reducing the number of accounting models for convertible debt instruments and convertible preferred shares. Limiting the accounting models results in fewer embedded conversion features being separately recognized from the host contract as compared with current GAAP. Convertible instruments that continue to be subject to separation models are (i) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting and (ii) convertible debt instruments issued with substantial premiums for which the premiums are recorded as additional paid-in capital. ASU 2020-06 also amends the guidance for the derivatives scope exception for contracts in an entity’s own equity to reduce form-over-substance-based accounting conclusions. ASU 2020-06 will be effective for the Company for fiscal years beginning after December 15, 2023. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020, including interim periods within those fiscal years. The Company is currently evaluating the potential impact adopting ASU 2020-06 will have on the Company’s consolidated financial statements and related disclosures.
F-14
| 3. | FAIR VALUE MEASUREMENTS |
The following are the major categories of assets measured at fair value on a recurring basis as of December 31, 2021 and 2020:
| As of December 31, 2021 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Assets: | ||||||||||||||||
| Short-term investments | $ | 33,248 | $ | — | $ | — | $ | 33,248 | ||||||||
| Total assets measured at fair value | $ | 33,248 | $ | — | $ | — | $ | 33,248 | ||||||||
| Liabilities: | ||||||||||||||||
| Derivative liability | $ | — | $ | — | $ | 5,379,878 | $ | 5,379,878 | ||||||||
| Warrant liability | — | — | 1,871,687 | 1,871,687 | ||||||||||||
| Total liabilities measured at fair value | $ | — | $ | — | $ | 7,251,565 | $ | 7,251,565 | ||||||||
| As of December 31, 2020 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Assets: | ||||||||||||||||
| Short-term investments | $ | 32,963 | $ | — | $ | — | $ | 32,963 | ||||||||
| Total assets measured at fair value | $ | 32,963 | $ | — | $ | — | $ | 32,963 | ||||||||
No transfers between levels have occurred in either reporting period presented.
| 4. | PREPAID EXPENSES AND OTHER CURRENT ASSETS |
Prepaid expenses and other current assets consist of the following:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Upfront research payments | $ | 554,878 | $ | 30,714 | ||||
| Goods and services tax receivable | 48,690 | 30,286 | ||||||
| Insurance | 32,853 | 27,335 | ||||||
| Dues and subscriptions | — | 16,863 | ||||||
| Consultants | 69,915 | 13,929 | ||||||
| License fee | 19,754 | — | ||||||
| Deposits | 6,839 | 7,818 | ||||||
| Miscellaneous | 4,387 | 6,077 | ||||||
| Total prepaid expenses and other current assets | $ | 737,316 | $ | 133,022 | ||||
F-15
| 5. | PROPERTY AND EQUIPMENT |
Property and equipment, net, consist of the following:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Laboratory equipment | $ | 66,403 | $ | 151,114 | ||||
| Computer equipment | 17,657 | 11,498 | ||||||
| Total property and equipment | 84,060 | 162,612 | ||||||
| Less: accumulated depreciation | (79,389 | ) | (84,501 | ) | ||||
| Property and equipment, net | $ | 4,671 | $ | 78,111 | ||||
Depreciation expense was $40,576 and $6,726 for the years ended December 31, 2021 and 2020, respectively. The Company recognized a gain on the sale of property and equipment of $59,157 for the year ended December 31, 2021.
| 6. | Intangible assets |
The Company has intangible assets consisting of acquired rights and patents with finite lives.
In March 2012, the Company acquired rights to a certain patented technology that it had licensed from its Chief Scientific Officer for C$100,000. The Company is amortizing this asset over its expected useful life of 15 years.
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Intangible assets | $ | 79,015 | $ | 78,417 | ||||
| Less: accumulated amortization | (51,401 | ) | (45,780 | ) | ||||
| Intangible assets, net | $ | 27,614 | $ | 32,637 | ||||
Amortization expense was $5,249 and $4,955 for the years ended December 31, 2021 and 2020, respectively.
As of December 31, 2021, the estimated expected amortization expense related to the Company’s intangible assets for each year through the year ended 2026 is $5,272 and, thereafter totals $1,254.
| 7. | ACCRUED LIABILITIES |
Accrued liabilities consist of the following:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Legal | $ | 171,777 | $ | — | ||||
| Accounting | 123,026 | 29,446 | ||||||
| Project work completed | 106,845 | 12,736 | ||||||
| Accrued interest | 54,398 | — | ||||||
| Annual meeting | 21,479 | 21,671 | ||||||
| Other | 42,568 | (17,652 | ) | |||||
| Accrued liabilities | $ | 520,093 | $ | 46,201 | ||||
| 8. | DEFERRED COMPENSATION |
The Company deferred cash payment of management compensation for the Executive Chairman. Chief Executive Officer, Chief Scientific Officer, Chief Medical Officer and Chief Development Officer at December 31, 2020, in the amount of $1,398,989. As of December 31, 2021, all deferred compensation to management was paid in full.
F-16
| 9. | Collaboration agreements |
In July 2020, the Company entered into two collaborative agreements (“BCNI Collaborations”) with BC Neuroimmunology Lab Inc. (“BCNI”) as follows:
Neurodegenerative Diseases
The Company and BCNI (“Neurodegen Collaboration”) agreed to develop and offer highly accurate and objective tests for detection, diagnosis and monitoring of AD. The operation will first offer existing blood-based assays for NfL and P-tau181. Further assays will be added, potentially incorporating the Company’s proprietary peptide antigens and tests for additional neurodegenerative diseases. The agreement was accounted for as a collaboration arrangement. Beginning in October 2020, each party contributed up to Canadian C$12,500 each month to cover operating costs up to the time that the operation becomes cashflow positive. The Company and BCNI acquired laboratory equipment that they jointly control. The Company contributed $19,973 and $115,900 during 2021 and 2020, respectively, of which $19,973 and $9,655 were recorded in research and development expenses in 2021 and 2020, respectively, in the accompanying consolidated statements of operations and comprehensive loss and $106,245 is reflected in property and equipment, net, in the accompanying consolidated balance sheets in 2020.
Covid-19
The Company and BCNI (“Covid-19 Collaboration”) agreed to provide the service of highly sensitive and specific serological assays for the detection and characterization of antibodies to the SARS-CoV-2 virus that is responsible for COVID-19. The Company and BCNI agreed to create an independent entity for the Covid-19 Collaboration to be established in 2021. The Company acquired 50% ownership interest in the future entity for $2,353. The Company recorded its investment in the future entity using the equity method of accounting and is reflected in other assets in the accompanying consolidated balance sheets. The Company is responsible for the funding of all operating expenses, with prior notification of the planned expenditures, to bring the assay through approval. The agreement was accounted for as a collaboration arrangement for the year ended December 31, 2020. The Company contributed $10,306 during 2020, which was recorded as research and development expense in 2020 on the accompanying consolidated statements of operations and comprehensive loss.
In January 2021, Covid-19 Collaboration became an independent entity. The Company and BCNI each owned 50% of the Covid-19 Collaboration. In February 2021, the Company funded C$25,000 of expenses, which would be paid back out of the profits, if any. As the Covid-19 Collaboration became an independent entity the Company accounted for it using the equity method. For the year ended December 31, 2021, the Company funded $77,549 of expenses. The Company recognized a pro-rata share of losses for the full amount of its investment in the Covid-19 Collaboration of $2,353 for the year ended December 31, 2021.
In December 2021, The BCNI Collaborations were terminated. The Covid-19 Collaboration redeemed the shares purchased by the Company for an aggregated redemption price of $2,353. A payment to the Company of C$128,000, which included the share redemption payment, and for the portion of the equipment purchased and related expenses incurred by the Company in relation to the Neurodegen Collaboration, was received by the Company on December 21, 2021.
| 10. | CONVERTIBLE DEBT |
In March 2021, the Company completed a $7.0 million private placement of Debentures. The Company allocated $3,567,442 of proceeds to the convertible debenture. The Company incurred $48,220 of issuance costs in connection with the private placement of which $24,575 was allocated to the debentures and amortized over the life of the debenture. The Debentures are convertible into common shares at the option of the holder at any time and from time to time at a conversion price of $0.10 and accrue interest at 1% per annum, which is payable annually. At the Company’s election, accrued interest may be paid in cash or common shares (such number of shares determined by dividing the interest due by the 5-day volume-weighted average price (“VWAP”) of the common shares). The Debentures mature on March 22, 2026. Prior to the maturity date, the Company is entitled to convert the Debentures at the conversion price upon raising an aggregate of $50 million in equity and/or debt. On the maturity date, the Company may redeem the outstanding principal amount of the Debentures in either cash or common shares (at the then 5-day VWAP less a 10% discount) or a combination thereof. The Company recognized $366,000 of interest expense relating to the amortization of the debt discount related to the derivative liability and issuance costs allocated to the debentures as of December 31, 2021.
The conversion feature has been recognized as a derivative liability recorded as a discount to the convertible debenture, adjusted to fair value each reporting period and being recorded in the consolidated statements of operations and comprehensive loss. The derivative liability has been valued at $3,432,558 at issuance date using a scenario-based valuation method using a Monte Carlo model, volatility of 101.43%, a risk-free interest rate of 0.15% and a selected debt yield of 15.96%. The derivative liability at December 31, 2021 has been valued at $5,379,878 using a scenario-based valuation method using a Monte Carlo simulation model, volatility of 95.95%, a risk-free interest rate of 1.15% and a selected debt yield of 15.96%. The total liability of the Debenture and the derivative liability at December 31, 2021 was $9,285,935. The portion of issuance costs allocated to the conversion feature of $23,645 were expensed when incurred in 2021.
| December 31, 2021 | ||||
| Balance at December 31, 2020 | $ | — | ||
| Derivative liability at issuance | 3,432,558 | |||
| Change in fair value of the derivative liability | 1,936,191 | |||
| Foreign exchange loss | 11,129 | |||
| Balance at December 31, 2021 | $ | 5,379,878 | ||
| 11. | COMMON SHARES |
The Company has authorized an unlimited number of both common and preferred shares. As of December 31, 2021 and 2020, the Company has 431,731,591 and 289,730,760 issued and outstanding common shares and no preferred shares as of December 31, 2021 and 2020, respectively. The common shares have no par value.
Common shares reserved for future issuance consists of the following:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Warrants | 93,644,288 | 37,165,711 | ||||||
| Convertible debt | 70,000,000 | — | ||||||
| Special Warrants | — | 32,439,162 | ||||||
| Options issued and outstanding under stock option plan | 44,282,249 | 38,771,749 | ||||||
| Deferred share units | 63,708 | 63,708 | ||||||
| Common shares available for grant under stock option plan | 16,907,820 | 10,387,190 | ||||||
| Total common shares reserved for future issuance | 224,898,065 | 118,827,520 | ||||||
F-17
The rights of the common shares are as follows:
Voting
Subject to any special voting rights or restrictions, holders of common shares entitled to vote shall have one vote per share.
Dividends
The Company’s board of directors may from time to time declare and authorize payment of dividends, if any, as they may deem advisable and need not give notice of such declaration to any shareholder. Subject to the rights of common shareholders, if any, holding shares with specific rights as to dividends, all dividends on common shares shall be declared and paid according to the number of such shares held and paid in Canadian dollars.
Liquidation Rights
In the event of the liquidation, dissolution or winding-up of the Company or any other distribution of the Company’s assets for the purpose of winding up the Company’s affairs, after the payment of dividends declared but unpaid, the holders of Common Shares shall be entitled pari passu to receive any remaining property of the Company.
Equity Transactions
In August 2021, the Company announced the closing of a public offering of 125,781,250 common share units at a price of US$0.16 per unit for gross proceeds of $20,125,000. The Company incurred $3,067,604 of share issuance costs in conjunction with the public offering. Each common share unit (“Unit”) consisted of one common share and one-quarter common share purchase warrant. Each whole warrant entitles the holder thereof to purchase one quarter common share at an exercise price of $0.21 per share at any time for five years. The warrants contain an acceleration clause allowing the Company to accelerate the expiry date of the warrants to 30 days following a time period during which the common share VWAP exceeds a TSX trading price of $0.63 for ten consecutive trading days.
The Company determined the allocation of the US$0.16 Unit issue price to the common shares and the one-quarter common share purchase warrants based on the relative fair values of the warrants, with the residual charged to equity. The common shares were allocated gross proceeds of $15,868,381 and share issue costs of $1,665,099. The common share warrants are accounted for as a warrant liability since the exercise price is in US$ while the Company’s functional currency is C$. The initial balance was calculated using the assumptions below resulting an allocation of gross proceeds of $2,739,221. Due to the existence of the acceleration option, the Company determined it was appropriate to fair value the warrants using a Monte Carlo Simulation model (“Monte Carlo”). The common shares issued were allocated a price of US$0.138 per share and the quarter common share purchase warrants were allocated a price of US$0.022. Assumptions used to determine the value of the common share warrants were: an average risk-free interest rate of 0.84%; annual volatility of 95.6%; and expected life of 5.0 years. The issuance costs allocated to the warrants based on the relative fair values of the warrants, amounted to $444,558 and were charged to general and administrative expense in the consolidated statements of operations and comprehensive loss.
As of December 31, 2021, the fair value of the warrants was calculated using the Monte Carlo model with the following parameters: risk free interest rate of 1.21%; annual volatility of 93.6%; and expected life of 4.7 years. The balance at December 31, 2021 was $1,871,687.
| December
31, 2021 | ||||
| Balance at December 31, 2020 | $ | — | ||
| Warrant liability at issuance | 2,739,221 | |||
| Change in fair value of the warrant liability | (840,555 | ) | ||
| Foreign exchange gain | (26,979 | ) | ||
| Balance at December 31, 2021 | $ | 1,871,687 | ||
Related to the sale of the Units, the Company paid certain intermediaries $1,408,750 and issued 8,804,687 compensation warrants. The compensation warrants are exercisable at any time for five years at an exercise price of USD$0.16 and do not have an acceleration clause. The compensation warrants have been issued as consideration for services provided by the intermediaries. The Company used a Black Scholes calculation to determine the fair value of the compensation warrants at the issuance date. The fair value of $957,947 was recorded to additional paid-in capital. Significant assumption used in the Black Scholes calculation included risk free interest rate of 1.21%; historical volatility of 95.6%; and a 5.0 year expiry.
F-18
| 12. | Warrants |
During February 2020, 4,790,251 warrants were exercised for total proceeds of $713,818 and 3,450,943 warrants expired.
During March 2020, the Company announced a repricing of various warrants to an exercise price of C$0.13, effective April 8, 2020 and expiring on May 22, 2020. The warrants repriced ranged in exercise prices of C$0.285 to C$0.48. At the end of the warrant repricing period, all unexercised warrants revert to the original exercise price. All other terms of the warrants shall remain unchanged. A total of 44,182,530 warrants were repriced, and, of the repriced warrants, 9,532,276 were exercised during the repricing period ending on May 22, 2020, for total proceeds of $894,272. In connection with the repricing, the Company recorded a fair value adjustment on the date of the modification of $85,005, which was treated as transaction costs and recorded to additional paid in capital.
In November 2020, the Company closed on a special warrant financing (“Special Warrants”). The Company issued 16,219,581 Special Warrants, resulting in proceeds of $1,483,531 ($1,256,828, net of issuance costs). Each Special Warrant is exercisable, without payment of any additional consideration by the holder, into one common share and one transferrable common share warrant (“Warrants”). Each Warrant entitles the holder to acquire one common share at an exercise price at C$0.20 per warrant share for 60 months until November 2025. Each Special Warrant will automatically convert at the earlier of the date that is (i) the third business day after a receipt for a final prospectus qualifying the distribution of the shares and warrants issuable upon the conversion of the Special Warrants and (ii) four months and one day after the issue date of the Special Warrants.
Related to the special warrant financing, the Company compensated certain intermediaries cash fees equal to 7% of the gross proceeds totaling $53,929 and issued 557,200 warrants, which have the same terms as the common share warrants.
In March 2021, the Special Warrants issued by the Company in connection with the November 2020 financing, automatically converted into 16,219,581 common shares and 16,219,581 common share warrants.
As at December 31, 2021, outstanding common share warrants and exercise prices denominated in C$ unless otherwise noted, related to unit offerings are as follows:
| Exercise Price $ | Number of Warrants | Expiry date | |||||
| 0.300 | 4,869,543 | August 2022 | |||||
| 0.285 | 1,265,010 | August 2022 | |||||
| 0.480 | 6,004,394 | April 2023 | |||||
| 0.480 | 8,379,500 | January 2024 | |||||
| 0.300 | 4,100,000 | June 2024 | |||||
| 0.300 | 9,049,066 | November 2024 | |||||
| 0.300 | 2,949,998 | December 2024 | |||||
| 0.200 | 16,776,781 | November 2025 | |||||
USD0.210 | 31,445,309 | August 2026 | |||||
USD0.160 | 8,804,687 | August 2026 | |||||
93,644,288 | |||||||
F-19
| 13. | SHARE-BASED COMPENSATION |
2007 Stock Option Plan
The Company maintains the 2007 Stock Option Plan (“2007 Option Plan”). In June 2015, the 2007 Option Plan was amended from a fixed option plan to a rolling share option plan pursuant to which the Company is authorized to grant options of up to 20% of its issued and outstanding common shares. Share options granted vest at various rates and have a term not exceeding ten years. As of December 31, 2021 and 2020, the Company had 16,844,112 and 10,387,190 options, respectively, available for grant under the 2007 Option Plan.
The following table summarizes the activity of the share options under the 2007 Option Plan for the years ended December 31, 2021 and 2020. All amounts are denominated in C$, except year and share amounts:
| Number
of Share Options | Weighted Average Exercise Price Per Share | Weighted Average Remaining Contractual Term (years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding as of January 1, 2020 | 38,323,749 | $ | 0.13 | 5.7 | $ | 2,855,797 | ||||||||||
| Granted | 550,000 | 0.19 | ||||||||||||||
| Forfeited | (102,000 | ) | 0.28 | |||||||||||||
| Outstanding as of December 31, 2020 | 38,771,749 | 0.13 | 4.8 | $ | 1,155,145 | |||||||||||
| Granted | 11,837,500 | 0.20 | ||||||||||||||
| Forfeited | (6,250,000 | ) | 0.24 | |||||||||||||
| Expired | (77,000 | ) | 0.30 | |||||||||||||
| Outstanding as of December 31, 2021 | 44,282,249 | 0.14 | 5.1 | $ | 2,231,293 | |||||||||||
| Vested and exercisable as of December 31, 2021 | 38,534,334 | $ | 0.13 | 4.4 | $ | 2,220,043 | ||||||||||
The aggregate intrinsic value of options outstanding, exercisable, and vested and exercisable is calculated as the difference between the exercise price of the underlying options, and the fair value of the Company’s common shares.
During the years ended December 31, 2021 and 2020, the Company granted share options with a grant date fair value of C$1,225,433 and C$78,347, respectively. During the years ended December 31, 2021 and 2020, there were no options exercised.
F-20
The fair value of the share options granted was estimated using Black Scholes with the following assumptions:
| Year Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Weighted average fair value of common shares | C$ | 0.10 | C$ | 0.14 | ||||
| Expected volatility | 92 | % | 105.0 | % | ||||
| Risk-free interest rate | 0.95 | % | 0.90 | % | ||||
| Expected dividend yield | – | % | – | % | ||||
| Expected term (years) | 4.5 | 5.5 | ||||||
Expected volatility is based on historical volatility of our shares over the expected life of the option, as our options are not readily tradable.
DSU Plan
The Company has a deferred share unit plan (“DSU Plan”) for senior officers. Under the DSU Plan, rights to the Company’s common shares may be awarded on a deferred payment basis up to a maximum of 1,000,000 common share units. Each common share unit will fully vest upon cessation of employment with the Company and then can be redeemed for one common share of the Company by the unitholder. The Company has 63,708 units outstanding as of December 31, 2021.
Share-based Payment Expense
The following table summarizes total share-based compensation included in the Company’s accompanying consolidated statements of operations and comprehensive loss:
| Year Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Research and development | $ | 144,905 | $ | 267,525 | ||||
| General and administrative | 370,724 | 86,877 | ||||||
| Total share-based compensation | $ | 515,629 | $ | 354,402 | ||||
As of December 31, 2021, there was $572,959 of unrecognized share-based compensation related to options outstanding, which were expected to be recognized over weighted-average remaining service period of 2.7 years.
| 14. | Income taxes |
As of December 31, 2021 and 2020, the net deferred tax assets have not been recognized in the accompanying consolidated financial statements. A valuation allowance is recognized to reduce the deferred tax asset as it is more likely than not that a tax benefit will not be realized.
The following are the significant components of the Company’s deferred taxes as of December 31:
| 2021 | 2020 | |||||||
| Non-capital losses carried forward | $ | 11,640,000 | $ | 8,981,000 | ||||
| Research and development expenditures | 3,421,000 | 3,230,000 | ||||||
| Investment tax credits | 2,201,000 | 2,078,000 | ||||||
| Tax value of technology rights and property and equipment in excess of accounting basis | 287,000 | 293,000 | ||||||
| Unrealized foreign exchange loss on convertible debt | 12,000 | — | ||||||
| Share issue costs | 550,000 | 138,000 | ||||||
| Total deferred income tax assets | 18,111,000 | 14,720,000 | ||||||
| Valuation allowance | (18,111,000 | ) | (14,720,000 | ) | ||||
| Net deferred income tax assets | $ | - | $ | - | ||||
F-21
As of December 31, 2021, the Company has available research and development expenditure credits for income tax purposes of approximately $12,911,000, which may be carried forward without expiration to reduce future taxable income.
As of December 31, 2021, the Company has non-capital income tax loss carry-forwards of approximately $43,936,000 available to reduce future income for income tax purposes. The income tax loss carry-forwards have expiry dates between the years 2026 and 2042.
As of December 31, 2021, the Company has approximately $2,890,000 of non-refundable investment tax credits available to offset future income taxes. The non-refundable investment tax credits have expiry dates between 2025 and 2035.
A reconciliation of the combined federal and provincial statutory income tax rate applied to the net loss for the year to the income tax recovery as of December 31 is as follows:
| 2021 | 2020 | |||||||
| Basic combined Canadian statutory income tax rate | 26.5 | % | 26.5 | % | ||||
| Income tax recovery based on statutory rate | $ | (2,457,000 | ) | $ | (1,126,000 | ) | ||
| Permanent differences | 396,000 | 94,000 | ||||||
| Share issue costs recorded, net of equity | (443,000 | ) | (61,000 | ) | ||||
| Unrecognized benefit of current year tax losses | 2,504,000 | 1,093,000 | ||||||
| $ | — | $ | — | |||||
The Company does not expect a significant change in the amount of unrecognized tax benefits over the next 12 months. However, any adjustments arising from certain ongoing examinations by tax authorities could alter the timing or amount of taxable income or deductions and these adjustments could differ from the amount accrued. The Company’s federal and provincial income tax returns files for all years remain subject to examination by the taxation authorities.
| 15. | Related party transactions |
During the years ended December 31, 2021 and 2020, the Company paid $322,639 and $231,354, respectively, for consulting services to a firm specializing in finance and strategic support for life science companies. The Chief Financial Officer of the Company is a managing director of the consulting firm.
In April 2016, the Company entered into a three-year, collaborative research agreement (“CRA”) with the University of British Columbia (“UBC”) and the Vancouver Coastal Health Authority in the amount of C$787,500, with the Company’s Chief Scientific Officer, as principal investigator at the UBC. In March 2018, the CRA was amended and funding was increased to C$892,500 over three years. In July 2018, the total funding commitment to UBC increased to C$1,130,000 over the period of the agreement. In February 2019, the CRA was amended, and funding was increased to C$2,130,000 for an additional two-year period. In September 2019, the CRA was amended, and funding was increased to C$2,630,000 for an additional one- year period. In November 2021, the CRA was amended for an additional grant of C$800,000 effective January 1, 2022, for the 2022 calendar year for total funding of C$3,430,000. During the years ended December 31, 2021 and 2020, the Company incurred costs of $393,341 and $353,253, respectively, and are included in research and development expenses in the accompanying consolidated statements of operations and comprehensive loss.
F-22
During the years ended December 31, 2021 and 2020, the Company paid $413,555 and $666,096, respectively, for management services to a company owned by the Company’s Chief Executive Officer and Executive Chairman for services rendered. The Company also reimbursed at cost the rental of an office, which is used by the Company. During the years ended December 31, 2021 and 2020, the Company incurred rental expense of $1,034 and $27,093, respectively, and are included in general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss.
| 16. | Commitments and contingencies |
Research, Development and License Agreements
The Company enters into research, development and license agreements with various parties in the ordinary course of business where the Company receives research services and rights to proprietary technologies. The agreements require compensation to be paid by the Company, typically, by a combination of the following:
| · | fees comprising amounts due initially on entering into the agreements and additional amounts due either on specified timelines or defined services to be provided; |
| · | milestone payments that are dependent on products developed under the agreements proceeding toward specified plans of clinical trials and commercial development; and |
| · | royalty payments calculated as a percentage of net sales, commencing on commercial sale of any product candidates developed from the technologies. |
Milestone and royalty related amounts that may come due under various agreements are dependent on, among other factors, preclinical safety and efficacy, clinical trials, regulatory approvals and, ultimately, the successful development and commercial launch of a new drug, the outcomes and timings of which are uncertain. Amounts due per the various agreements for milestone payments will accrue once the occurrence of a milestone is likely. Amounts due as royalty payments will accrue as commercial revenues from the product are earned. Through December 31, 2021, no events have occurred that require accrual of any milestone or royalty related amounts.
UBC and the Vancouver Coastal Health Authority Agreement
In April 2016, the Company entered into a three-year, CRA with the UBC and the Vancouver Coastal Health Authority. The agreement was amended various times through September 2019. Refer to Note 15 Related Party Transactions.
UBC Agreement
In February 2009, the Company entered into an agreement with UBC to further the development and commercialization of certain technology developed, in part, by the Company’s Chief Scientific Officer. The agreement was amended and restated in October 2015. Under the amended and restated agreement, the Company is committed to make royalty payments based on revenue earned from the licensed technology. An annual license fee is payable over the term of the agreement. The agreement remains effective unless terminated under the provisions of the agreement. Through December 31, 2021 no accruals for royalty payments have been made.
University Health Network Agreement
In April 2006, an additional amendments through November 2013, the Company entered into an agreement with the University Health Network, Toronto, to license certain technology and related intellectual property. Under the agreement, the Company is committed to make milestone payments of up to C$635,000 based on the successful outcomes of clinical and regulatory outcomes, buyout payments and royalty payments based on revenue earned from the licensed technology. As of December 31, 2021 and 2020, no accruals for any milestones or royalty payments have been made.
F-23
Indemnification
In the ordinary course of business, the Company enters into agreements that may include indemnification provisions. Pursuant to such agreements, the Company may indemnify, hold harmless and defend an indemnified party for losses suffered or incurred by the indemnified party. Some of the provisions will limit losses to those arising from third party actions. In some cases, the indemnification will continue after the termination of the agreement. The maximum potential amount of future payments the Company could be required to make under these provisions is not determinable. The Company has never incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. The Company has also entered into indemnification agreements with its directors and officers that may require the Company to indemnify its directors and officers against liabilities that may arise by reason of their status or service as directors or officers. The Company currently has directors’ and officers’ insurance.
Leases
During the years ended December 31, 2021 and 2020, the Company made short-term lease payments in the amount of $20,806 and $19,071, respectively, and are included in general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss. The Company’s commitment for future payments under its lease agreements is C$8,765 for the year ended December 31, 2022.
| 17. | NET LOSS PER SHARE |
The following table sets forth the computation of basic and diluted net loss per share attributable to common shareholders:
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Numerator: | ||||||||
| Net loss attributable to common shareholders | $ | 9,790,106 | $ | 4,250,280 | ||||
| Denominator: | ||||||||
Weighted-average shares outstanding used in computing net loss per share attributable to common shareholders, basic and diluted | 347,137,045 | 285,599,827 | ||||||
| Net loss per share attributable to common shareholders, basic and diluted | $ | (0.03 | ) | $ | (0.01 | ) | ||
The following outstanding potentially dilutive common shares equivalents were excluded from the computation of diluted net loss per share for the periods presented because including them would have been antidilutive:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Options issued and outstanding under stock option plan | 44,282,249 | 38,771,749 | ||||||
| Warrants | 93,644,288 | 37,165,711 | ||||||
| Convertible debt | 70,000,000 | — | ||||||
| Special warrants | — | 32,439,162 | ||||||
| Deferred share units | 63,708 | 63,708 | ||||||
| Total | 207,990,244 | 108,440,330 | ||||||
F-24
| 18. | Subsequent events |
In January 2022, the UBC CRA was amended, and funding was increased to C$5,030,000 for an additional two years. This amendment, along with the November 2021 amendment extends the project for an additional three years, effective January 1, 2022.
F-25
Appendix A
ProMIS Neurosciences Inc. Patent Portfolio Summary
Patent / Publication Type of IP / Jurisdiction |
Title / Inventor(s) / Assignee (s) | Status / Patent Term / Comments |
Utility - Canada
3004593 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
National Phase of PCT/CA2016/051306
COMPUTER IMPLEMENTED METHODS |
Utility - United States
US 2018-0330045 A1 2018-11-15
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
National Phase of PCT/CA2016/051306
COMPUTER IMPLEMENTED METHODS
|
3374906 2022-01-05
Utility - Belgium
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS |
3374906 2022-01-05
Utility - Denmark
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS |
3374906 2022-01-05
Utility - France
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS |
3374906 2022-01-05
Utility - Germany
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS |
3374906 2022-01-05
Utility - Switzerland
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS |
3374906 2022-01-05
Utility - United Kingdom
16863269.3 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
Based on EP 16863269.3, which is a National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS
|
Utility - Hong Kong
19101704.4 2016-11-09
|
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
Based on CN Application 201680065612.6
COMPUTER IMPLEMENTED METHODS
|
Utility - India
201817021051 2016-11-09 |
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
National Phase of PCT/CA2016/051306
COMPUTER IMPLEMENTED METHODS
|
6952351 2021-09-30
Utility - Japan
2018-522708 2016-11-09 |
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
National Phase of PCT/CA2016/051306
Calculated Term Expiration: 2036-11-09
COMPUTER IMPLEMENTED METHODS
|
Utility - South Korea
10-2018-7015912 2016-11-09 |
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
National Phase of PCT/CA2016/051306
COMPUTER IMPLEMENTED METHODS
|
Utility - China
201680065612.6 2016-11-09 |
SYSTEMS AND METHODS FOR PREDICTING MISFOLDED PROTEIN EPITOPES BY COLLECTIVE COORDINATE BIASING
Steven Samuel Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
PENDING
National Phase of PCT/CA2016/051306
COMPUTER IMPLEMENTED METHODS
|
Utility - Canada
3142040 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
COMPUTER IMPLEMENTED METHODS
|
Utility - United States
17/614487 2020-05-27
*not yet published |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - Australia
2020284288 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - China
202080052086.6 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - Europe
20815021.9 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - India
202147059454 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - Japan
To be assigned 2020-05-27 |
CONFORMATION-SPECIFIC EPITOPES IN TAU, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Steven Plotkin Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2020/050722 IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A TAUOPATHY
|
Utility - Canada
3148562 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - United States
17/283292 2019-10-07
*not yet published |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - Australia 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - Europe
19870641.8 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - India
202127020718 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - Japan
2021-516452 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - Hong Kong
62022047684.5 2019-10-07 |
CONFORMATION-SPECIFIC EPITOPES IN ALPHA-SYNUCLEIN, ANTIBODIES THERETO AND METHODS RELATED THEREOF
Neil R. Cashman Steven S. Plotkin Xubiao Peng Joanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
PENDING
National Phase of PCT/CA2019/051434
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING OR TREATING A SYNUCLEINOPATHY
|
Utility - Canada
3064785 2018-05-30 |
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2018/050634
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING TDP-43
|
11214613 2022-01-04
Utility - United States
US 2020-0165327 A1 2020-05-28 |
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA |
ISSUED
National Phase of PCT/CA2018/050634
Calculated Term Expiration: 2038-05-30
ANTIBODIES, METHOD OF DETECTING TDP-43 |
Utility - United States
17/541812 2021-12-03
*not yet published |
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA
|
PENDING
Divisional of 16/616832
TDP-43 IMMUNOGENS |
Utility - Europe
18810255.2 2018-05-30 |
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2018/050634
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING TDP-43
|
Utility Hong Kong
62020014330.8 2018-05-30
|
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2018/050634
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING TDP-43
|
Utility - Japan
2019-565224 2018-05-30 |
EPITOPES IN THE RNA RECOGNITION MOTIF 1 (RRM1) OF TDP-43 AND MISFOLDING-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin Xubiao Peng
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2018/050634
IMMUNOGENS, ANTIBODIES, METHOD OF DETECTING TDP-43
|
Utility - Canada
3123116 2019-12-16 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - United States
US 2022-0056118 A1 2022-02-24 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - Australia
2019399680 2019-12-16 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - Europe
19897296.0 2019-12-16 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - India
202127031355 2019-12-16 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - Japan
2021-533492 2019-12-16 |
ANTIBODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2019/051823
ANTIBODIES, IMMUNOGENS AND METHODS OF TREATMENT AND DETECTION OF MISFOLDED TDP-43 |
Utility - PCT
PCT/CA2021/050597 2021-04-29 |
SINGLE CHAIN ANTIBODIES AND INTRABODIES TO MISFOLDED TDP-43 AND METHODS OF USE
Neil R. Cashman Johanne Kaplan Beibei Zhao
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Published
TDP-43 NUCLEIC ACIDS AND METHODS OF TREATMENT
|
Utility - PCT
PCT/CA2021/050521 2021-04-16 |
COMPOSITIONS AND METHODS FOR INHIBITING TDP-43 AND FUS AGGREGATION
Neil R. Cashman Steven S. Plotkin Beibei Zhao Ching-Chung Hsueh Catherine M. Cowan
THE UNIVERSITY OF BRITISH COLUMBIA |
Published
ANTI-SENSE OLIGONUCLEOTIDES AND COMPOSITIONS AND METHODS OF TREATMENT |
2009301580 2016-03-10
Utility - Australia
2009301580 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
2,743,361 2021-05-25
Utility - Canada
2,743,361 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
2342220 2021-05-12
Utility - Germany
09818707.3 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Granted
Based on EP 09818707.3, which is a National Phase of PCT/CA2009/001413
METHODS FOR PREDICTING AND MAKING IMMUNOGENS
Calculated Term Expiration: 2029-10-06
|
2342220 2021-05-12
Utility - Denmark
09818707.3 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Granted
Based on EP 09818707.3, which is a National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
2342220 2021-05-12
Utility - France
09818707.3 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Granted
Based on EP 09818707.3, which is a National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
2342220 2021-05-12
Utility - United Kingdom
09818707.3 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Granted
Based on EP 09818707.3, which is a National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
2342220 2021-05-12
Utility - Netherlands
09818707.3 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Granted
Based on EP 09818707.3, which is a National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
5898495 2016-03-11
Utility - Canada
2011529431 2009-10-06 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
National Phase of PCT/CA2009/001413
Calculated Term Expiration: 2029-10-06
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
10,475,525 2019-11-12
Utility - United States
US 2016-0132633 A1 2016-05-12 |
METHODS AND SYSTEMS FOR PREDICTING MISFOLDED PROTEIN EPITOPES
Neil R. Cashman Steven S. Plotkin William C. Guest
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
Divisional of 12/574637 ( now abandoned)
Calculated term expiration: 2030-09-05
METHODS FOR PREDICTING AND MAKING IMMUNOGENS |
Utility - Canada
3004498 2015-11-09
|
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES
|
10759837 2020-09-01
Utility - United States
US 2018-0346535 A1 2018-12-06 |
ANTI-AMYLOID BETA ANTIBODIES BINDING TO A CYCLIC AMYLOID BETA PEPTIDE
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued National Phase of PCT/CA2016/051305
Calculated term expiration: 2036-11-09
Terminal Disclaimer: US 10,751,382
A-BETA ANTIBODIES |
Utility - United States
US 2021-0087243 A1 2021-03-25 |
ANTI-AMYLOID BETA ANTIBODIES BINDING TO A CYCLIC AMYLOID BETA PEPTIDE
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
Continuation of 15/774,778
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES
|
Utility - Australia
2016353553 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Allowed
National Phase of PCT/CA2016/051305
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Australia
NYA |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Unfiled Divisional of AU 2016353553
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - China
201680065514.2 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Europe
16863268.5 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Hong Kong
19101690.0 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - India
201817020841 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Japan
2018-522803 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - South Korea
10-20187015910 2016-11-09 |
AMYLOID BETA EPITOPES AND ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051305 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Canada
3004482 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
10772969 2020-09-15
Utility - United States
US 2018-0326086 A1 2018-11-15 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued National Phase of PCT/CA2016/051300
Calculated term expiration: 2036-11-09
Terminal Disclaimer: US10,751,382 |
Utility - United States
US 2021-0154315 A1 2021-05-27 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending Continuation of 15/774,805
IMMUNOGENS, METHODS OF DETECTING A-BETA DISEASES AND MAKING A-BETA ANTIBODIES |
Utility - Australia
20163535552 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Allowed National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Australia
NYA |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Unfiled Divisional of AU 20163535552 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - China
201680065463.3 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Europe
16863264.4 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Hong Kong
19101691.9 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending Based on CN 201680065463.3 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - India
201817020836 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Japan
2018-522707 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - South Korea
10-2018-7015908 2016-11-09 |
N-TERMINAL EPITOPES IN AMYLOID BETA AND CONFORMATIONALLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending National Phase of PCT/CA2016/051300 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Canada
3004494 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
10774120 2020-09-15
Utility - United States
US 2018-0319856 A1 2018-11-08 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
National Phase of PCT/CA2016/051303
Calculated term expiration: 2036-11-09
Terminal Disclaimer: 10,751,382
A-BETA ANTIBODIES |
Utility - United States
US 2021-0087244 A1 2021-03-25 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
Continuation of 15/774,707
IMMUNOGENS, METHODS OF TREATING A-BETA DISEASES |
Utility - Australia
2016354688 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Allowed
National Phase of PCT/CA2016/051303
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Australia
2022201737 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
Divisional of AU 2016354688
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - China
201680065488.3 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Europe
16863266.9 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Hong Kong
19101692.8 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - India
201817020839 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Japan
2018-523030 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303
IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - South Korea
10-2018-7015909 2016-11-09 |
EPITOPES IN AMYLOID BETA MID-REGION AND CONFORMATIONALLLY-SELECTIVE ANTIBODIES THERETO
Neil R. Cashman Steven S. Plotkin
THE UNIVERSITY OF BRITISH COLUMBIA |
Pending
National Phase of PCT/CA2016/051303 IMMUNOGENS, ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Canada
3031135 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - United States
US 2020-0172602 A1 2020-06-04 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
METHODS OF INHIBITING AND A-BETA AND TREATING A-BETA DISEASES |
Utility - Australia
2017299858 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - China
201780043930.7 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Europe
17830149.5 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Hong Kong
19131617.3 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - India
201917005362 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Japan
2019-501965 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - South Korea
10-2019-7004028 2017-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2017/050866
ANTIBODIES, METHODS OF DETECTING OR TREATING A-BETA DISEASES |
Utility - Canada
3070085 2018-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2018/050875
ANTIBODIES, COMPOSITIONS AND METHODS OF DETECTING OR TREATING A-BETA OLIBOMER DISEASES |
Utility - United States
US 2020-0181247 A1 2020-06-11 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2018/050875
ANTIBODIES, COMPOSITIONS AND METHODS OF DETECTING OR TREATING A-BETA OLIBOMER DISEASES |
Utility - Europe
18835520 2018-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2018/050875
ANTIBODIES, COMPOSITIONS AND METHODS OF DETECTING OR TREATING A-BETA OLIBOMER DISEASES |
Utility - Hong Kong
620200078306 2018-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2018/050875
ANTIBODIES, COMPOSITIONS AND METHODS OF DETECTING OR TREATING A-BETA OLIBOMER DISEASES |
Utility - Japan
2020-502655 2018-07-18 |
ANTIBODIES TO AMYLOID BETA
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
National Phase of PCT/CA2018/050875
ANTIBODIES, COMPOSITIONS AND METHODS OF DETECTING OR TREATING A-BETA OLIBOMER DISEASES |
10751382 2020-08-25
Utility - United States
US 2019-0151401 A1 2019-05-23 |
METHODS AND COMPOSITIONS FOR PREVENTING AND TREATING A-BETA OLIGOMER-ASSOCIATED AND/OR -INDUCED DISEASES AND CONDITIONS
Neil R. Cashman
THE UNIVERSITY OF BRITISH COLUMBIA |
Issued
Continuation of 15/808842 (abandoned) which is a CIP of PCT/CA2017/050866 and PCT/2016/051305 and PCT/2016/051303 and PCT/2016/051300
Calculated term expiration: 2036-11-09
Terminal Disclaimer: US 10,774,120 and US 10,759,837
A-BETA ANTIBODIES AND COMPOSITIONS |
Utility - United States
US 2021-0038678 A1 2021-02-11 |
METHODS AND COMPOSITIONS FOR PREVENTING AND TREATING A-BETA OLIGOMER-ASSOCIATED AND/OR -INDUCED DISEASES AND CONDITIONS
Neil R. Cashman Steven S. Plotkin Johanne Kaplan Judith Maxwell Silverman Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Pending
Continuation of 16/148601
IMMUNOGEN COMPOSITIONS AND METHOD OF TREATING A-BETA OLIGOMER DISEASES |
Utility - PCT
PCT/CA2021/050431 2021-03-31 |
ANTIBODIES TO MISFOLDED AMYLOID BETA
Neil R. Cashman Johanne Kaplan Ebrima Gibbs
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Published
ANTIBODIES TO, METHODS OF DETECTING OR TREATING A-BETA OLIGOMER DISEASES |
10,053,510 2018-08-21
Utility - United States
US 2016-0215055 A1 2016-07-28 |
FasR ANTIBODIES FOR DIAGNOSTIC AND THERAPEUTIC USE
Marni Diane Uger Veronica Ciolfi Neil R. Cashman
THE UNIVERSITY OF BRITISH COLUMBIA PROMIS NEUROSCIENCES INC. |
Issued
National Phase of PCT2014/000457
Calculated term expiration: 2034-07-03
Patent Term Adjustment: 38 days
ANTIBODIES, PHARMACUETICAL COMPOSITIONS AND METHODS OF DETECTING MISFOLDED FasR |
2642848 2016-07-12
Utility- Canada
2642848 2007-03-05
|
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. UNIVERSITY HEALTH NETWORK |
ISSUED
National Phase of PCT/CA2007/000346
Calculated Term Expiration: 2027-03-05
USE FOR TREATING A SOD1 MEDIATED NEURODEGENERATIVE CONDITION |
7977314 2011-07-12
Utility - United States
US 20070292410-A1 2007-03-05 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman
PROMIS NEUROSCIENCES INC. |
ISSUED
Continuation in part of 11/565,967 (USP779469) and 11/367609 (USP 7439324) and US filing corresponding to PCT/CA2007/000346
Calculated Term Expiration: 2028-01-22
Patent Term Adjustment: 690 days
METHODS OF ALS TREATMENT |
7887803 2011-02-15
Utility - United States
US-2008-02206251-A1 2007-12-20 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman
PROMIS NEUROSCIENCES INC. |
ISSUED
Continuation in Part of 11/682,217 and 11/565967 (USP779469) and 11/367609 (USP 7439324)
Calculated Term Expiration: 2027-02-22
Patent Term Adjustment: 357 days
METHOD OF ALS TREATMENT |
8709422 2014-04-29
Utility - United States
US-2011-0212097-A1 2011-09-01 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. |
ISSUED
Continuation of 11/850,502
Calculated Term Expiration: 2026-03-03
Terminal Disclaimer US 8,778,885
Patent Term Adjustment: 95 days
METHOD OF ALS TREATMENT |
9637552 2017-05-02
Utility - United States
US-2014-0348822-A1 2014-11-27 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. |
ISSUED
Continuation of 13/155939 (USP 8778885)
Calculated Term Expiration: 2024-08-20
Terminal Disclaimers: US 7,763,710; US 8,075,891; US 7,439,324; US 8,513,387; US 7,977,314; US8,778,885; US7,887,803; and 8,709,422
METHOD OF TREATING SOD-1 NEURODEGENERATIVE DISEASE |
5823663 2015-10-16
Utility - Japan
2008-556626 2007-03-05 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. UNIVERSITY HEALTH NETWORK |
ISSUED
National Phase of PCT/CA2007/000346
Calculated Term Expiration: 2027-03-05
MEDICAMENTS, PHARMACEUTICAL COMPOSITIONS FOR TREATING SOD-1 NEURODEGENERATIVE DISEASE |
2514823 2018-05-02
Utility - United Kingdom
07710682.1 2007-03-05 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. UNIVERSITY HEALTH NETWORK |
Issued
Based on EP 07710682.1 which is a National Phase of PCT/CA2007/000346
Calculated Term Expiration: 2027-03-05
ANTIBODIES AND IMMUNOGENS FOR TREATING ALS |
2514823 2018-05-02
Utility - Germany
07710682.1 2007-03-05 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. UNIVERSITY HEALTH NETWORK |
Issued
Based on EP 07710682.1 which is a National Phase of PCT/CA2007/000346
Calculated Term Expiration: 2027-03-05
ANTIBODIES AND IMMUNOGENS FOR TREATING ALS |
2514823 2018-05-02
Utility - France
07710682.1 2007-03-05 |
METHODS AND COMPOSITIONS TO TREAT AND DETECT MISFOLDED-SOD1 MEDIATED DISEASES Neil R. Cashman Avijit Chakrabartty Joachim Berhhard Ostermann Rishi Rakhit
PROMIS NEUROSCIENCES INC. UNIVERSITY HEALTH NETWORK |
Issued
Based on EP 07710682.1 which is a National Phase of PCT/CA2007/000346
Calculated Term Expiration: 2027-03-05
ANTIBODIES AND IMMUNOGENS FOR TREATING ALS |
2536305 2015-10-27
Utility - Canada
2536305 2004-08-20 |
SOD-1 EPITOPES AND ANTIBODIES
Neil R. Cashman Marty Letho
PROMIS NEUROSCIENCES INC. |
ISSUED
National Phase of PCT/CA2004/001503
Calculated Term Expiration: 2024-08-20
SOD1 IMMUNOGEN, ANTIBODY, COMPOSITION, METHOD OF MAKING AND DETECTION AGENT |
7439324 2008-10-21
Utility - United States
US 2006-0246517 A1 2006-03-03 |
ALS-SPECIFIC PEPTIDE COMPOSITION
Neil R. Cashman
PROMIS NEUROSCIENCES INC. |
Issued
CIP of PCT/CA2004/001503
Calculated term expiration: 2024-12-11
Patent Term Adjustment: 113 days
SOD-1 IMMUNOGEN COMPOSISITON |
8,075,891 2011-12-13
Utility - United States
US-2011-0135673-A1 2011-06-09 |
ANTIBODIES THAT BIND ALS SPECIFIC EPITOPES AND METHODS OF MAKING
Neil R. Cashman
PROMIS NEUROSCIENCES INC. |
Issued
Continuation of 12/236,731 (USP7763710)
Calculated term expiration: 2024-08-20
SOD-1 ANTIBODIES |
9,523,697 2016-12-20
Utility - United States
US-2011-0124018-A1 2011-05-26 |
DETECTION OF PATHOGENIC ABETA USING AN EPITOPE PROTECTION ASSAY
Neil R. Cashman Marty Lehto
PROMIS NEUROSCIENCES INC. |
Issued
Continuation of 10/568,729 (Abandoned), which is a National Phase of PCT/CA2004/001503)
Calculated term expiration: 2025-06-14
Patent Term Adjustment: 298 days
METHODS FOR DETECTING A-BETA |
9,625,476 2017-04-18
Utility - United States
US-2014-0335538-A1 2014-11-13 |
METHODS OF DIAGNOSING ALS
Neil R. Cashman
PROMIS NEUROSCIENCES INC. |
Issued
Continuation of 13/313,869 (USP 8828389)
Calculated Term Expiration: 2024-08-20
(Terminal disclaimer US 8,075,891; US 8,778,885; and US 8,828,389)
ALS DIAGNOSTIC METHODS |
5357111 2013-09-06
Utility - Japan
2010-265290 2010-07-05 |
EPITOPE PROTECTION ASSAY AND METHOD FOR DETECTING PROTEIN CONFORMATIONS
Neil R. Cashman Marty Lehto
PROMIS NEUROSCIENCES INC. |
Issued
Divisional of 2006-523496
Calculated Term Expiration: 2024-08-20
SOD-1 IMMUNOGENS AND ANTIBODIES |
8,513,387 2013-08-20
Utility - United States
US-2011-0020358-A1 2011-01-27 |
Methods and Compositions for Detecting Amyotrophic Lateral Sclerosis
Avijit Chakrabartty Rishi Rakhit Neil Cashman
PROMIS NEUROSCIENCES INC. |
Issued
Continuation of 11/565967 (USP7794692)
Calculated Term Expiration: 2027-07-12
(Terminal Disclaimer: US 7,794,692)
(Patent term adjustments: 224 days)
SOD1 ANTIBODY AND METHODS OF MAKING |
2,877,505 2020-09-22
Utility - Canada
2,877,505 2013-06-11 |
ANTIBODIES AND CONJUGATES THAT TARGET MISFOLDED PRION PROTEIN
Marni Diane Uger Viengthong Chai Veronica Ciolfi Neil R. Cashman Wah Yau Wong Heman Lap-Man Chao Baomin Tian
HELIX BIOPHARMA CORP. PROMIS NEUROSCIENCES INC. |
Issued
National Phase of PCT/CA2013/000569
Calculated Term Expiration: 2033-06-11
PRION PROTEIN ANTIBODIES, PHARMACEUTICAL COMPOSITIONS AND USES |
6430373 2018-11-09
Utility - Japan
2015-516391 2013-06-11 |
ANTIBODIES AND CONJUGATES THAT TARGET MISFOLDED PRION PROTEIN
Marni Diane Uger Viengthong Chai Veronica Ciolfi Neil R. Cashman Wah Yau Wong Heman Lap-Man Chao Baomin Tian
HELIX BIOPHARMA CORP. PROMIS NEUROSCIENCES INC. |
Issued
National Phase of PCT/CA2013/000569
Calculated Term Expiration: 2033-06-11
PRION PROTEIN ANTIBODIES, PHARMACEUTICAL COMPOSITIONS AND USES |
9,296,819 2016-03-29
Utility - United States
US-2015-0166668-A1 2015-06-18 |
ANTIBODIES AND CONJUGATES THAT TARGET MISFOLDED PRION PROTEIN
Marni Diane Uger Viengthong Chai Veronica Ciolfi Neil R. Cashman Wah Yau Wong Heman Lap-Man Chao Baomin Tian
HELIX BIOPHARMA CORP. PROMIS NEUROSCIENCES INC. |
Issued
National Phase of PCT/CA2013/000569
Calculated Term Expiration: 2033-06-11
PRION ANTIBODY, COMPOSITION, METHODS OF DETECTING AND TREATING PRION DISEASES
|
SIGNATURES
Pursuant to the requirements of Section 12 of the Securities Exchange Act of 1934, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized.
| PROMIS NEUROSCIENCES INC. | |
| By: Eugene Williams | |
| Title: Chairman and Chief Executive Officer |
Date: April 4, 2022
122
EXHIBIT INDEX
| Exhibit No. | Description of Exhibit | |
| 3.1# | Articles | |
| 3.1.1# | Certificate of Amendment to the Articles | |
| 3.2# | By-laws | |
| 3.2.1# | By-law No. 2 | |
| 4.1# | Shareholder Rights Plan Agreement dated January 22, 2016, Amended and Extended as of May 15, 2019, by and between ProMIS Neurosciences Inc. and Computershare Trust Company of Canada as Rights Agent. | |
| 4.2# | Unsecured Convertible Debenture dated March 22, 2021. | |
| 10.1#+ | Joint Venture Agreement dated July 7, 2020 by and between ProMIS Neurosciences Inc. and BC Neuroimmunology Lab Inc. | |
| 10.2#+ | Joint Venture Agreement dated July 8, 2020 by and between ProMIS Neurosciences Inc. and BC Neuroimmunology Lab Inc. | |
| 10.3#+ | Collaborative Research Agreement by and between The University of British Columbia and Provincial Health Services Authority (on behalf of Children’s & Women’s Health Centre of British Columbia Branch, a public hospital) and ProMIS Neurosciences Inc. effective April 1, 2016. | |
| 10.3.1#+ | Amendment No. 1 dated February 13, 2017 to the Collaborative Research Agreement by and between The University of British Columbia and Provincial Health Services Authority (on behalf of Children’s & Women’s Health Centre of British Columbia Branch, a public hospital) and ProMIS Neurosciences Inc. effective April 1, 2016. | |
| 10.3.2#+ | Amendment No. 2 dated July 5, 2018 to the Collaborative Research Agreement by and between The University of British Columbia and Provincial Health Services Authority (on behalf of Children’s & Women’s Health Centre of British Columbia Branch, a public hospital) and ProMIS Neurosciences Inc. effective April 1, 2016. | |
| 10.3.3#+ | Amendment No. 3 dated February 13, 2019 to the Collaborative Research Agreement by and between The University of British Columbia and Provincial Health Services Authority (on behalf of Children’s & Women’s Health Centre of British Columbia Branch, a public hospital) and ProMIS Neurosciences Inc. effective April 1, 2016. | |
| 10.3.4#+ | Amendment No. 4 dated September 9, 2019 to the Collaborative Research Agreement by and between The University of British Columbia and Provincial Health Services Authority (on behalf of Children’s & Women’s Health Centre of British Columbia Branch, a public hospital) and ProMIS Neurosciences Inc. effective April 1, 2016. | |
| 10.4#+ | Amended and Restated License Agreement dated October 6, 2015 by and between The University of British Columbia and ProMIS Neurosciences Inc. | |
| 10.5#+ | License Agreement dated August 3, 2006 by and between Amorfix Life Sciences Ltd. and an Affiliate of Biogen Idec Inc. | |
| 10.6#+ | Exclusive License Agreement dated July 14, 2010 by and between Amorfix Life Sciences Ltd. and Biogen Idec MA Inc. | |
| 10.7#+ | License Agreement dated April 4, 2006 by and between University Health Network and Amorfix Life Sciences Inc. | |
| 10.7.1# | Amendment dated July 13, 2006 to the License Agreement dated April 4, 2006 by and between University Health Network and Amorfix Life Sciences Inc. |
123
| 10.7.2#+ | Amendment No. 2 dated July 11, 2007 to the License Agreement dated April 4, 2006 by and between University Health Network and Amorfix Life Sciences Ltd. | |
| 10.7.3#+ | Amendment No. 3 dated November 4, 2013 to the to the License Agreement dated April 4, 2006 by and between University Health Network and Amorfix Life Sciences Ltd. | |
| 10.8#†+ | Consulting Agreement dated April 1, 2021 by and between Elliot Paul Goldstein, MD and ProMIS Neurosciences Inc. | |
| 10.8.1*†+ | Consulting Agreement dated October 1, 2021 by and between Elliot Goldstein, MD and ProMIS Neurosciences Inc. | |
| 10.9#†+ | Advisory Consulting Agreement dated May 26, 2021 by and between ProMIS Neurosciences Inc. and David Wishart. | |
| 10.10#†+ | Consulting and Advisory Agreement dated March 1, 2005 by and between Amorfix Life Sciences Ltd. And Neil Cashman. | |
| 10.11#†+ | Consulting Agreement dated June 29, 2015 by and between Amorfix Life Sciences Ltd. and Virtua, LLC. | |
| 10.12#†+ | Consulting Agreement dated October 17, 2016 by and between ProMIS Neurosciences Inc. and Danforth Advisors, LLC. | |
| 10.12.1#†+ | Amendment No. 1 dated March 27, 2017 to Consulting Agreement dated October 17, 2016 by and between ProMIS Neurosciences Inc. and Danforth Advisors, LLC. | |
| 10.12.2#† | Amendment No. 2 dated December 12, 2017 to Consulting Agreement dated October 17, 2016 by and between ProMIS Neurosciences Inc. and Danforth Advisors, LLC. | |
| 10.12.3#† | Amendment No. 3 dated August 28, 2018 to Consulting Agreement dated October 17, 2016 by and between ProMIS Neurosciences Inc. and Danforth Advisors, LLC. | |
| 10.12.4#†+ | Amendment No. 4 dated March 27, 2017 to Consulting Agreement dated October 17, 2016 by and between ProMIS Neurosciences Inc. and Danforth Advisors, LLC. | |
| 10.13# | Form of Finder’s Warrant Certificate dated April 30, 2018. | |
| 10.14# | Form of Non-US Warrant Certificate dated April 30, 2018. | |
| 10.15# | Form of Employee Stock Option Commitment. | |
| 10.16#+ | Form of Unit Subscription Agreement for Non-U.S. Subscribers dated February 25, 2020. | |
| 10.17#+ | Form of Unit Subscription Agreement for Non-U.S. Subscribers dated June 17, 2019. | |
| 10.18#+ | Form of Unit Subscription Agreement for U.S. Subscribers dated November 27, 2018. | |
| 10.19#+ | Form of Unit Subscription Agreement for U.S. Subscribers dated October 21, 2019. | |
| 10.20#+ | Form of Unit Subscription Agreement for U.S. Subscribers dated April 13, 2018. | |
| 10.21#+ | Form of Unit Subscription Agreement for Non-U.S. Subscribers dated April 13, 2018. | |
| 10.22#+ | Form of Unit Subscription Agreement for Non-U.S. Subscribers dated November 27, 2018. | |
| 10.23#+ | Form of Unit Subscription Agreement for Non-U.S. Subscribers dated October 21, 2019. | |
| 10.24#+ | Form of Finder’s Warrant Certificate dated November 2020. | |
| 10.25#+ | Form of Non-U.S. Finder’s Warrant Certificate dated January 2019. | |
| 10.26#+ | Form of Non-U.S. Warrant Certificate dated January 2019. | |
| 10.27#+ | Form of Non-U.S. Warrant Certificate dated June 2019. | |
| 10.28#+ | Form of U.S. Warrant Certificate dated January 2019. | |
| 10.29#+ | Form of U.S. Warrant Certificate dated November 2020. | |
| 10.30# | Form of Special Warrant Certificate dated November 4, 2020. | |
124
| 10.31# | Form of U.S. Special Warrant Certificate dated November 4, 2020. | |
| 10.32#+ | Form of Warrant Certificate dated November 2020. | |
| 10.33#+ | Technology License Agreement dated February 1, 2006 by and between Dr. Neil Roy Cashman and Amorfix Life Sciences Ltd. | |
| 10.34#+ | Service Agreement dated September 1, 2020 by and between The University of Saskatchewan and ProMIS Neurosciences Inc. | |
| 10.35#+ | Assignment Agreement dated February 18, 2005 by and between Neil R. Cashman and Marty Lehto and the Governing Council of the University of Toronto and Amorfix Life Sciences Ltd. | |
| 10.35.1# | Amendment Agreement dated April 1, 2005 to the Assignment Agreement dated February 18, 2005 by and between Neil R. Cashman and Marty Lehto and the Governing Council of the University of Toronto and Amorfix Life Sciences Ltd. | |
| 10.36#†+ | Executive Employment Agreement of Eugene Williams dated December 31, 2021. | |
| 10.37#†+ | Executive Employment Agreement of Gavin Malenfant dated December 31, 2021. |
| 10.38#† | ProMIS Neruosciences Inc. 2015 Stock Option Plan. | |
| 10.39#† | Amorfix Life Sciences Ltd. Deferred Share Unit Plan for Canadian Senior Officers. | |
| 10.40* | Form of Non-U.S. Finder’s Warrant Certificate dated November 2019. | |
| 10.41* | Form of Non-U.S. Warrant Certificate dated November 2019. | |
| 10.42* | Form of U.S. Warrant Certificate dated November 2019. | |
| 10.43* | Form of Non-U.S. Warrant Certificate dated November 2020. | |
| 10.44* | Form of Broker Warrant dated August 2021. | |
| 10.45* | Form of Non-U.S. Warrant Certificate dated August 2021. | |
| 10.46* | Form of U.S. Warrant Certificate dated August 2021. |
| * | Filed herewith. | |
| # | Previously filed. | |
| † | Indicates a management contract or compensatory plan or arrangement. | |
| + | Certain identified information has been excluded from the exhibit pursuant to Item 601(a)(6) and/or Item 601(b)(10)(iv) of Regulation S-K. | |
125